 2017, Vol. 47
2017, Vol. 47

2. 陕西延长石油有限责任公司, 陕西 西安 710069
2. Shaanxi Yanchang Petroleum Company, Xi′an 710069, China
氢能是一种极具发展潜力的清洁能源, 具有燃烧热值高、燃烧产物无污染等优点。目前, 制氢方法主要包括:醇类制氢[1-2]、烃类制氢[3-4]以及氨分解制氢[5-9]等。使用烃类和醇类等含碳燃料重整制氢会伴随着CO或者CO2的生成, 用于燃料电池时极易引起电极中毒, 碳氧化物后期分离也将增加制氢成本。氨气不仅具有高的储氢密度(氢的质量分数17.6 %), 而且分解产物不含碳氧化物, 同时, 氨气易液化便于储存和运输, 在制氢上具有诸多优势, 因而氨分解制氢受到学者越来越多的关注。氨分解催化制氢[10-14]通常在高温下进行, 而且贵金属催化剂[13-14]的使用进一步增加了氨分解制氢的经济投入, 不利于氨分解制氢的发展。基于此,等离子体技术在氨分解制氢方面有广泛的前景。等离子体是物质的第四态, 能够在充分活化反应物的同时降低能耗。1980年, Agostino[8]报道了射频等离子体应用于氨分解的研究, 并从动力学角度对等离子体条件下氨分解的机理进行了探究。颜士鑫[15]用滑弧放电等离子体在常温常压条件下对于氨分解进行了探索, 当供电电压为10 000 V, 氨气流速为6 L/min时, 氨气的转化率可达12 %。赵越[6]采用交流弧放电等离子体用于氨分解的研究, 考察了不同电极对于氨分解反应影响。在输入功率为24.3 W时, Cu电极下氨气的转化率约为43 %而Ni电极能达到97 %。王丽[16]联合介质阻挡放电等离子体和不同催化剂用于氨分解制氢, 结果表明,在450℃时, 联合等离子体和Co/SiO2催化剂时氨气的转化率可达99 %。
本文将负电晕放电等离子体用于氨分解反应。本课题组曾将负电晕技术用于二氧化碳和氨气生成附加产物尿素[17], 以及资源化处理二氧化碳和碘蒸气成四碘甲烷[19]。负电晕放电能够激发并活化电负性气体, 电负性气体分子能够捕获自由电子而形成负离子。负离子化的气体分子具有更高的能量, 能够产生更激烈分子间的碰撞。本文采用负电晕放电和商用铁基催化剂相结合的方法用于氨分解制氢, 并对反应机理进行初步探究。
1 实验部分 1.1 等离子体反应器结构负电晕放电反应装置如图 1所示。电晕放电针由上端放入反应装置, 另一头连接高压放电电源, 使其通过放电针尖产生负电晕放电。电晕放电针是金属银材质, 每根针长约35 mm, 直径约0.16 mm。反应器设有催化剂塔板, 用来装填催化剂。

|
1 高压输入电源;2 接管口;3 进气口;4 温控装置;5 催化剂塔板;6 出气口 图 1 负电晕放电反应装置图 Fig. 1 The diagram of the negative corona discharge reaction equipment |
SJ-2000E型高压输入电源由广东佛山顺德有限公司生产, 用于产生直流电且最高电压为15 kV。本研究在此基础上增加了一套半波整流单元用来整流输出电压为负压, 使其能够稳定的产生直流负高压。
1.2 反应性能评价采用商用氨合成铁基催化剂(主要成分为Fe3O4), 催化剂用量为0.50 g。首先, 催化剂在氩气保护下, 以10℃/min的速率升温至550℃, 后氢气还原2 h, 并在氩气保护下降至反应温度。稳定30 min后通氨气, 同时开启负电晕放电装置。
FULI 9790型气相色谱用于气相产物的检测, 采用GDX-01填充柱, 柱温为50℃, 进样器温度为100℃, 检测器温度为130℃。色谱尾气经稀酸溶液吸收并测得流量后排空。固定床式加热炉由上海实验仪器有限公司生产, 可程序升温用于调控反应体系温度, 最高温度可达900℃。
反应性能评价包括氨气转化率η, 氢气收率Y和协同效应因子F。
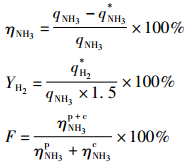
|
式中:qNH3为氨气进口流量, q*NH3为氨气出口流量, q*H2为氢气出口流量。ηP+CNH3,ηPNH3,ηCNH3分别代表等离子体和催化剂联合、单独等离子体、单独催化剂下氨气的转化率。
1.3 催化剂表征XRD测试在Rigaku D/Max-2500PC型X衍射仪上进行, 管电压为40 kV, 管电流为40 mA, 扫描范围为10°~90°。NH3-TPD采用Quantachrome公司生产的CBT-01型动态化学吸附仪进行检测。将50 mg样品装于石英管中, 先在200℃氦气气氛下吹扫60 min, 除去表面的吸附物种, 再降温至50℃, 注入NH330 min后经氦气吹扫30 min除去物理吸附的NH3后以10℃/min的速度升温到800℃, 氦气气速为40 mL/min。
2 结果与讨论 2.1 放电功率, 气速和温度对等离子体氨分解的影响 2.1.1 输入功率对等离子体氨分解的影响图 2为常温下, 氨气气速为30 mL/min时放电输入功率对氨分解的影响。输入功率增加, 氨气的转化率逐渐增加。这是由于氨气的转化率与电子的平均能量密切相关。电子的平均能与E/n值(即与电场强度E成正比和反应物浓度n成反比)相关[20]。当反应物浓度n不变时, 电场强度随着输入功率的增加而增加从而使E/n值变大, 电子的平均动能增加, 活性粒子增加, 反应物分子在更为激烈的碰撞中发生裂解, 氨气的转化率增加。与此同时, 氨气分解产生氢气的收率也随着放电功率的增加而增加。

|
图 2 常温下放电输入功率对氨分解的影响 Fig. 2 The effect of discharge input power on the decomposition of ammonia at ambient temperature |
常温、输入功率为480 W时, 随着气速的增加, 氨气的转化率降低(如图 3), 生成的氢气将减少, 收率降低。这是由于气速的增加反应气体在反应区域的停留时间变短, 导致大部分气体分子离开放电区域时没有完全活化。而负电晕等离子体在活化反应物分子时, 活性粒子的寿命短, 在气速增加时, 活性粒子不能发生有效碰撞而分解, 产生的氢气量也随之减少。因此, 气速增加时氨气转化率降低, 氢气收率也降低。

|
图 3 常温下氨气体积流量对氨分解的影响 Fig. 3 The effect of ammonia flow rate on the decomposition of ammonia at ambient temperature |
由图 4可以看出,氨气键为30mL/min,在低输入功率下(96 W,192 W), 温度从100℃增加到260℃, 氨气转化率逐渐增加, 而在高输入功率下(288~480W), 氨气转化率从100~260℃先增加后保持不变。这是由于在输入功率较低时等离子的活化能量不足以完全激发和活化反应物分子, 这时温度在反应中起主导作用。因为温度增加, 分子平均动能增加, 分子间的碰撞加剧使得氨气转化率增加。而在高的输入功率时, 等离子体能够充分活化反应物分子, 这时温度对反应体系的影响不大, 等离子体起主导作用。

|
图 4 温度对等离子体氨分解的影响 Fig. 4 The effect of temperature on the decomposition of ammonia |
如图 5(a)和图 5(b), 在480 W,氨气气速为30mL/min时,加入催化剂后, 温度从100℃增加到260℃, 氨气的转化率和氢气的收率都有所提高。图 4表明, 等离子体和催化剂结合时协同效应因子大于1, 说明二者产生了协同效应。并且, 协同效应因子随着温度增加而增加。而且, 在温度低于260℃时单独采用Fe基催化剂催化时转化率小于5 %, 说明Fe基催化剂在低温条件下活性低。而当等离子体输入能量不变时, 温度增加能够提高反应物分子的动能, 从而使得更多的反应物分子被活化而增加协同效应。

|
图 5 温度对氨分解的影响 Fig. 5 The effect of temperature on the decomposition of ammonia |
如图 6(a)和图 6(b)所示, 260℃时, 反应气速为30mL/min,输入功率增加氨气的转化率提高, 氢气收率也随之增加。同时,协同效应因子随输入功率增加而逐渐增加。这是由于随着输入功率的增加, 投入到等离子体区域的能量增加, 将会有更多的NH3分子被激发。在负电晕条件下, NH3分子捕获电子而形成氨负离子, 一方面, 氨负离子携带的能量高, 活化的NH3将会有更高的动能, 在放电区域能够产生更为激烈的碰撞而发生分解;另一方面, 动能更高的氨负离子将更容易运动到催化剂表面, 进一步在催化剂表面发生反应。

|
图 6 放电输入功率对氨分解的影响 Fig. 6 The effect of discharge input power on the decomposition of ammonia |
对反应前后催化剂进行了XRD表征, 结果如图 7, 反应前催化剂出现的衍射峰归属为金属态的铁。放电反应后的催化剂除金属铁的衍射峰, 还出现了Fe的氮化物(FeNx,Fe2N)的衍射峰。但是单独催化剂催化反应后只出现了Fe2N的衍射峰, 不存在Fe的衍射峰。等离子体条件下NH3在分解为N2和H2的同时会产生一些表面氮原子,而等离子体条件下, 催化剂表面的N原子更容易脱附, 因而可以保持金属态Fe的存在[17-18]。金属态Fe是氨气分解真正的活性中心, 在单独催化剂催化过程中, 铁基催化剂会被氮化而形成氮化铁, 形成的氮化铁占据了铁催化剂的活性位从而使得催化剂活性降低。但是,放电反应能够抑制铁基催化剂表面形成氮化铁, 使铁基催化剂保持活性。

|
图 7 反应前后Fe基催化剂的XRD图 Fig. 7 The XRD diagram of Fe based catalysts before and after reaction |
对反应前后的催化剂进行了NH3-TPD分析, 结果如图 8, 还原后的铁基催化剂有两个氨气的脱附峰, 而等离子体处理后的催化剂表面的氨气在313℃,528℃和625℃脱附。而在单催化反应后的催化剂表面氨气脱附偏移到660℃, 说明单催化反应后氨气的脱附更困难, 催化剂活性降低。这也说明氮化物的形成会降低催化活性, 导致氨分解的转化率降低。

|
图 8 反应前后Fe基催化剂的NH3-TPD图 Fig. 8 The NH3-TPD diagram of Fe based catalysts before and after reaction |
负电晕等离子体可以激发电负性分子, 而产生活性负离子。由放电针释放的电子携带的能量为4~5 eV, 在放电时, NH3可以形成NH2-负离子(电子亲和力0.77 eV)[21]。如下所示。
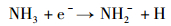
|
(1) |

|
(2) |
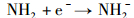
|
(3) |
随后, 氨负离子进一步发生自由基反应。其中, H2的产生于式(4),(5),(7)和(8)的自由基反应。在式(6)和(7)的反应中可以产生氮原子, 部分氮原子经过式(9)过程形成N2分子, 另一部分氮原子吸附在铁基催化剂表面形成氮化物。
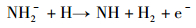
|
(4) |

|
(5) |
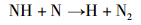
|
(6) |
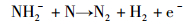
|
(7) |
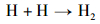
|
(8) |
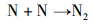
|
(9) |
由此可见, 氨气负离子的形成过程对于氨分解有着非常重要的作用。而在低温下催化剂对于氨气分子的吸附量少, 进而催化剂表面的自由基反应将大大减少。当负电晕联合铁基催化剂时, 氨气在负电晕条件下将会被激发活化, 形成的氨气负离子能够在催化剂表面充分地进行自由基反应。结合催化剂表征分析结果, 等离子体和催化剂协同效应产生于两个方面。一方面, 等离子体可以充分促进反应物分子的活化, 使氨气分子形成氨气负离子。另一方面, 等离子参与反应时可以抑制催化剂表面氮化物的形成。因此, 在等离子体结合催化剂催化氨分解时, 等离子体起着非常重要的作用。
3 结语常温下, 单独等离子催化氨分解时氨气转化率和氢气收率随着输入功率的增加而增加, 随着气速的增加而降低。并且, 氨气转化率随温度增加变化不大, 尤其在输入功率较高的情况下。在等离子体和催化剂结合催化反应时, 提高了氨气的转化率和氢气的收率产生了协同效应。260℃时, 氨气转化率随输入功率增加而增加。随着温度提高而增加。
催化剂表征表明Fe基催化剂在催化时会被氮化, 而负电晕放电反应中, 不仅能够激发和活化反应物分子, 同时能够有效抑制催化剂表面氮化物的形成, 而保持催化剂的高活性, 使得催化剂和等离子体之间产生协同效应, 因此等离子体协同催化剂是一种催化氨分解制氢的有效手段。该方法也可以推广应用于其他热力学不可行反应, 具有很好的应用前景。
| [1] |
DU C M, MA D Y, WU J. Plasma-catalysis reforming for H2 production from ethanol[J]. International Journal of Hydrogen Energy, 2015, 40(45): 15398-15410. DOI:10.1016/j.ijhydene.2015.09.096 |
| [2] |
ZHANG H, LI X D, BOA Z. Non-oxidative decomposition of methanol into hydrogen in a rotating gliding arc plasma reactor[J]. International Journal of Hydrogen Energy, 2015, 40(46): 15901-15912. DOI:10.1016/j.ijhydene.2015.09.052 |
| [3] |
MURADOV N Z, VEZIROGLU T N. From hydrocarbon to hydrogen-carbon to hydrogen economy[J]. International Journal of Hydrogen Energy, 2005, 30(3): 225-237. DOI:10.1016/j.ijhydene.2004.03.033 |
| [4] |
SPATH P L, MANN M K. Life cycle assessment of hydrogen production via natural gas steam reforming[J]. Natural Gas Steam Reforming National Renewable Energy Laboratory, 2000, 2: 1-33. |
| [5] |
QIU H, MARTUS K, LEE W Y. Hydrogen generation in a microhollow cathode discharge in high-pressure ammonia-argon gas mixtures[J]. International Journal of Mass Spectrometry, 2014, 233(1-3): 19-24. |
| [6] |
ZHAO Y, WANG L, ZHANG J L, et al. Decomposition of ammonia by atmospheric pressure AC discharge: Catalytic effect of the electrodes[J]. Catalyst Today, 2013, 211: 72-77. DOI:10.1016/j.cattod.2013.03.027 |
| [7] |
MILLER G P, BAIRD J K. Radio frequency plasma decomposition of ammonia: a comparison with radiation chemistry using the G value[J]. Journal of Physical Chemistry, 1993, 97(42): 10984-10988. DOI:10.1021/j100144a015 |
| [8] |
AGOSTINO R D, CRAMAROSSA F, BENEDICTIS S D. Kinetic and spectroscopic analysis of NH3 decomposition under Radio Frequency Plasma at moderate pressures[J]. Plasma Chemistry and Plasma Processing, 1981, 1(1): 19-35. DOI:10.1007/BF00566373 |
| [9] |
SILVA H, NIELSEN M G, FIORDALISO E M. Synthesis and characterization of Fe-Ni/-Al2O3 egg-shell catalyst for H2 generation by ammonia decomposition[J]. Applied Catalysis A General, 2015, 152(2): 548-556. |
| [10] |
OKURA K, OKANISHI T, MUROYAMA H. Promotion effect of rare-earth elements on the catalytic decomposition of ammonia over Ni/Al2O3 catalyst[J]. Applied Catalysis A General, 2015, 505: 77-85. DOI:10.1016/j.apcata.2015.07.020 |
| [11] |
PODILA S, DRISS H, ZAMAN S F. Hydrogen generation by ammonia decomposition using Co/MgO-La2O3 catalyst: Influence of support calcination atmosphere[J]. Journal of Molecular Catalysis A Chemical, 2016, 414: 130-139. |
| [12] |
PODILA S, ALHAMED Y A, ALZAHRANI A A. Hydrogen production by ammonia decomposition using Cocatalyst supported on Mg mixed oxide systems[J]. International Journal of Hydrogen Energy, 2015, 40(45): 15411-15422. DOI:10.1016/j.ijhydene.2015.09.057 |
| [13] |
YIN S F, XU B Q, NG C F. Nano Ru/CNTs: a highly active and stable catalyst for the generation of COx-free hydrogen in ammonia decomposition[J]. Applied Catalysis B Environmental, 2004, 48(4): 237-241. DOI:10.1016/j.apcatb.2003.10.013 |
| [14] |
LI X K, JI W J, ZHAO J, et al. Ammonia decomposition over Ru and Ni catalysts supported on fumed SiO2, MCM-41, and SBA-15[J]. Journal of Catalysis, 2005, 236(2): 181-189. DOI:10.1016/j.jcat.2005.09.030 |
| [15] |
颜士鑫, 李晓东, 钟犁. 滑弧放电等离子体分解氨气制氢[J]. 燃料与科学技术, 2011, 17(2): 186-190. |
| [16] |
WANG L, YI Y H, GUO H C. NH3 decomposition for H2 generation: Effects of cheap metals and supports on plasma-catalyst synergy[J]. Journal of Catalysis, 2015, 5: 4167-4174. |
| [17] |
XIANG X F, GUO L, MA X Y. Urea formation from carbon dioxide and ammonia at atmospheric pressure[J]. Environmental Chemistry Letters, 2012, 10(3): 295-300. DOI:10.1007/s10311-012-0366-2 |
| [18] |
王丽.等离子体催化剂氨分解制氢协同效应研究[D].大连: 大连理工大学, 2014. http://cdmd.cnki.com.cn/Article/CDMD-10141-1013197388.htm
|
| [19] |
XIANG X F, GUO L, MA X Y. Reduction of carbon dioxide into tetraiodomethane at 1atm[J]. Environmental Chemistry Letters, 2012, 10(4): 413-418. DOI:10.1007/s10311-012-0379-x |
| [20] |
MOK Y S, LEE H J, DORS M. Improvement in selective catalytic reduction of nitrogen oxides by using dielectric barrier discharge[J]. Chemical Engineering Journal, 2005, 110(1-3): 79-85. |
| [21] |
CHEN J, DAVIDON J H. Model of the negative DC corona plasma: comparison to the positive DC corona plasma[J]. Plasma Chem Plasma Process, 2003, 23(1): 83-102. DOI:10.1023/A:1022468803203 |