 2017, Vol. 47
2017, Vol. 47
 , 李稳宏1
, 李稳宏1 2. 西北有色金属研究院技术部,陕西西安 710016;
3. 中国人民解放军 第451医院, 陕西 西安 710054
2. Department of Technology, Northwest Institute for Non-ferrous Metal Research, Xi′an 710016, China;
3. The No.451 Hospital, Chinese People′s Liberation Army, Xi′an 710054, China
加氢催化剂失活分为中毒失活、结焦失活和烧结失活[1-4]。失活催化剂需再生后重复使用, 减少催化剂成本和环境污染。加氢精制催化剂性质各异, 使用周期和工艺条件不同, 因此催化剂再生时, 要区别对待[5]。
本文针对自制且失活的煤焦油加氢精制催化剂, 根据热重分析确定精制剂的再生条件, 利用XRD,BET,XPS,XRF检测手段研究了催化剂再生前后物化性质的变化。利用模型化合物喹啉(QI)及二苯并噻吩(DBT)对比了新鲜和再生精制剂的活性,为催化剂的失活再生, 重复利用提供参考。
1 实验部分 1.1 加氢精制催化剂的制备 1.1.1 试剂与仪器拟薄水铝石比表面积≥250m2/g, 孔容≥0.3mL/g, Na2O质量分数≤0.3%。HY-分子筛硅铝比>5, Na2O质量分数≤0.2%。聚丙烯酰胺、田箐粉、硝酸、磷酸、乙二胺四乙酸二钠(EDTA)、硝酸镍、偏钨酸铵均为分析纯。
101-0A型鼓风干燥箱, SX2-10-12型箱式电阻炉。
1.1.2 载体制备取一定量SA-拟薄水铝石, 加入适量HY-分子筛、聚丙烯酰胺、田箐粉, 搅拌均匀后, 加入无机酸(硝酸+磷酸)混合水溶液, 在双螺杆挤条机上混碾挤压, 采用直径1.5mm的圆型孔板挤条成型。经120℃烘箱干燥6h, 600℃马弗炉焙烧4h, 制得催化剂载体。
1.1.3 催化剂制备实验用等体积浸渍法制备加氢精制催化剂, 首先测定干燥后载体的单位吸水量。配制相应体积的EDTA、硝酸镍、偏钨酸铵的混合浸渍液。将干燥后的催化剂载体浸渍到混合液中8h, 在120℃干燥6h, 550℃焙烧4h, 得到加氢精制催化剂。
1.2 催化剂分析测试方法热重分析用DTU-2A型热分析仪, 60mL/min空气, 10℃/min升温速率; 比表面积及孔隙度分析在TriStar Π 3020M吸附仪上进行, 吸附温度77K, 抽真空速率10℃/min, 平衡时间60min, 脱气温度200℃; 物相分析采用D/Max 2500型X-射线衍射仪, 扫描范围3°~90°, 步进宽度0.02°/步, CuKα射线, 石墨单色电极; 表面元素价态采用ESCALab220I-XL型多功能X射线光电子能谱仪, 激发源为Al KαX射线, 功率300 W; 元素分析采用日本理学Rigaku 3271E型X射线荧光光谱仪。
1.3 催化剂活性评价实验用质量分数分别为4%二苯并噻吩、2%喹啉的甲苯溶液作为反应物。采用100mL高压反应釜, 反应物装量20g, 催化剂装量5g。模型化合物加氢实验条件比较温和, 反应温度320℃, 压力4MPa, 氢油体积比800:1, 反应时间4h。反应前对催化剂进行预硫化。
原料及产物硫氮含量由KY-3000SN型微量硫氮测定仪测得。用色谱-质谱仪(GC-MS)分析加氢产物的组成[6], GC 2060型气相色谱仪进行定量分析, FID检测器, HP-5非极性毛细色谱柱, 定量方法为外标法。
2 结果和讨论 2.1 热重分析用热重分析结果确定催化剂的再生温度[7]。图 1为失活催化剂的热重(TG)和差热(DTA)曲线。

|
图 1 失活催化剂TG-DTA谱图 Fig. 1 TG-DTA profile of deactivated catalyst |
在图 1TG曲线上, 分别在室温~250 ℃,250~360℃和360~576℃区间有4.53%,2.13%,7.46%的失重, 最大失重处温度分别为220℃,338℃和527℃。在失活催化剂的DTA曲线上, 室温~ 250℃左右存在吸热过程, 338℃,527℃处有2个较强的放热峰, 632℃处有个较弱的放热峰。失活催化剂TG曲线上室温~250℃区间的失重属于表面物理吸附水、未抽涤干净的轻组分油脱附以及纤维状积炭的总体贡献。失活催化剂表面硫化态金属氧化过程失重较少, 对应TG曲线上250~360℃的失重和DTA曲线上338℃处的放热峰, 放热较大。失活催化剂表面假石墨型积炭致密, 不易被氧化, 它与DTA曲线上527℃的热峰及TG曲线上360~576℃区间的失重相对应。DTA曲线上632℃处的放热峰归结为催化剂表面金属发生结晶转变所致。
综上所述, 为尽可能烧掉催化剂上的积碳而又不让催化剂的晶体结构发生烧结, 确定催化剂的再生温度为550℃。催化剂再生过程存在放热现象, 因此要有适度的升温速率, 0~250℃升温速度为5℃/min, 250~360℃升温速率为3℃/min, 360~550℃升温速率为2℃/min, 550℃恒温时间为4h。
2.2 再生前后催化剂物化性质变化1) BET测定
新鲜及再生前、后催化剂的孔结构数据见表 1。
|
|
表 1 催化剂孔结构结果 Tab. 1 The results of pore characterization |
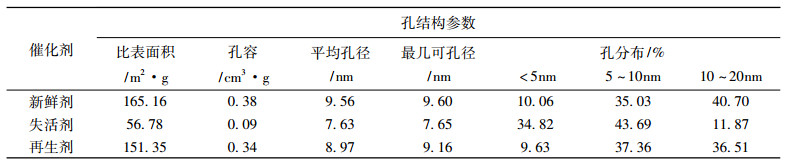
由表 1知, 失活催化剂表面及孔隙生成大量积碳, 使催化剂的比表面积和孔容大幅降低。再生后催化剂比表面积恢复率为91.64%, 孔容恢复率为89.47%, 孔径恢复率为95.42%, 孔结构得到有效恢复。
2) XRF测定
对新鲜及再生前后催化剂的组成进行了分析, 结果见表 2。
|
|
表 2 催化剂组成分析 Tab. 2 The compositions of catalysts |
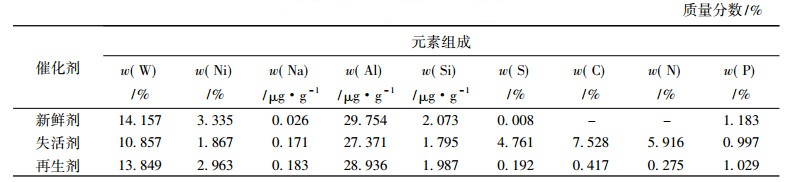
从表 2看出, 失活催化剂表面积炭严重。再生后, C,N及S的烧除率分别为94.46%,95.35%及95.97%。这说明催化剂上的积碳在再生过程中基本烧掉, 但仍有少量C,N等杂质存在。再生后催化剂上的金属含量较新鲜剂低, 一是金属流失所致, 另一方面是催化剂上残存的金属杂质和S,C等引起的数据测定偏低。
3) XRD测定
对新鲜及再生前后催化剂进行物相分析, XRD谱图见图 2。

|
A Fresh; B Deactivated; C Regenerated 图 2 催化剂XRD谱图 Fig. 2 XRD pattern of catalysts |
可以看出, 新鲜催化剂只在46°和67°左右(400)和(440)晶面存在归属于γ-Al2O3的特征衍射峰, 活性金属在载体表面分散性好。再生催化剂在27°出现归属于Al(WO4)3的衍射峰, 但峰形小且宽, 说明晶粒尺寸小, 活性金属分散均匀。
4) XPS测定
为比较新鲜及再生催化剂预硫化后表面金属赋存形态, 对催化剂进行宽能量范围扫描检测。结果见图 3。

|
A Fresh; B Regenerated 图 3 催化剂的XPS全范围谱图 Fig. 3 XPS spectra of catalysts |
从XPS全谱可见, 再生后催化剂的表面元素变化不大, 由于反应中催化剂的积碳、金属沉积、活性组分硫化等因素的影响, 再生后的催化剂表面出现了S,N以及部分金属杂质, 部分元素含量发生了变化, 这与前文中的元素分析结果一致。
为了进一步分析再生催化剂的活性组分赋存形态, 分别对W和Ni进行窄区扫描(W 4f,Ni 2p)。
1) Ni 2p谱图分析

|
图 4 再生催化剂Ni 2p XPS谱图 Fig. 4 XPS spectra of Ni 2p for regenerated catalyst |
|
|
表 3 再生催化剂的Ni 2p拟合结果 Tab. 3 Fitting results of Ni 2p for regenerated catalyst |
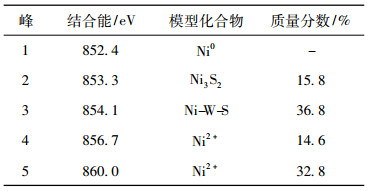
由此可知, 再生后催化剂Ni 2p各组分状态均合理存在, 拟合图谱与原始谱图基本重合。并且,催化剂中Ni主要存在形式为NiS和Ni-W-S, 约占到组分总量的85%(质量分数)左右, 其次为硫化不充分的Ni0和Ni3S2[8-9]。
2) W 4f谱图分析

|
图 5 再生催化剂W 4f XPS谱图 Fig. 5 XPS spectra of W 4f for regenerated catalyst |
|
|
表 4 再生催化剂的W 4f XPS拟合结果 Tab. 4 Fitting results of W 4f for regenerated catalyst |
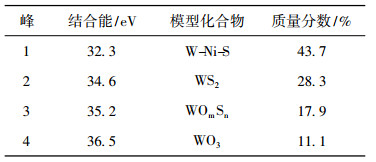
由此可知, W 4f各组分状态均合理存在, 拟合图谱与原始谱图基本重合, 催化剂中W主要存在形式为32.3eV左右的Ni-W-S及34.6eV左右的WS2[10-12], 其总量为82%(质量分数)左右。催化剂再生后硫化度较好, 其中的硫氧化钨存在的原因是硫化不完全或是硫化后重新被氧化所致。
2.3 活性评价加氢反应物为4%(质量分数)二苯并噻吩、2%(质量分数)喹啉的甲苯溶液, 用反应脱硫、脱氮率及产物分布来考察再生催化剂的精制效果, 新鲜及再生催化剂加氢精制效果见表 5。
|
|
表 5 加氢精制效果 Tab. 5 Effect of hydrorefining |
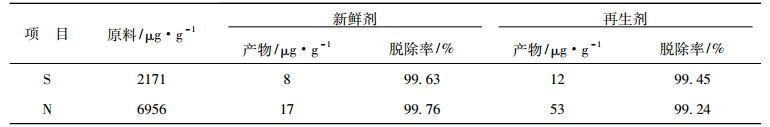
由此可知, 失活催化剂再生后, 硫、氮脱除率分别下降0.18%和0.52%, 但二者脱除率仍然在99%以上, 催化剂活性基本上得到恢复。
经GC-MS分析, 确定加氢产物种类, 并采用GC-FID对产物进行定量分析。计算得新鲜及再生催化剂加氢处理结果见表 6。
|
|
表 6 催化剂对二苯并噻吩及喹啉加氢处理性能 Tab. 6 Catalytic performance in hydroprocessing of benzothiophene and quinoline |
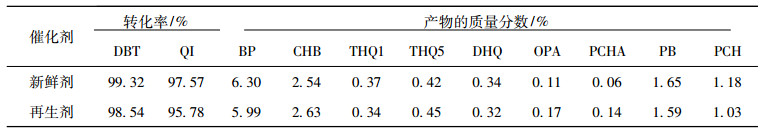
由此可知, 二苯并噻吩(DBT)的加氢产物主要有联苯(BP)和少量苯基环己烷(CHB)。BP是DBT分子先加氢生成二氢产物, 然后断开C—S键得到, CHB是DBT分子的一个芳环先加氢饱和, 再断开C—S键, 脱除H2S[13]。与新鲜催化剂相比, 再生催化剂对DBT的转化率及加氢产物分布影响不大。
喹啉(QI)在加氢反应中首先发生一个芳香环的加氢饱和, 然后是C—N键的断裂和脱氮反应或是进一步加氢饱和再脱氮, 反应条件苛刻[14]。由此可知, QI加氢产物复杂[15], 有1, 2, 3, 4-四氢喹啉(THQ1), 5, 6, 7, 8-四氢喹啉(THQ5), 邻丙基苯胺(OPA), 2-丙基环己苯胺(PCHA), 丙基环己烷(PCH), 丙苯(PB), 其中PCH和PB为不含氮产物。再生催化剂加氢反应中, QI转化率变化不大, 但是PB和PCH的产量稍有下降, 而OPA和PCHA的产量增加, 说明再生剂的脱氮选择性降低, 这一结果也正是再生剂脱氮率降低的原因。同时,QI加氢脱氮反应中存在OPA加氢脱氮生成PB和PCHA加氢脱氮生成PCH的两条平行脱氮反应。
3 结论1) 采用热重分析可以确定失活催化剂的再生条件, 使失活催化剂恢复活性。
2) 催化剂再生后比表面积回复率为91.64%, 孔容恢复率为89.47%, 平均孔径回复率为95.42%;催化剂再生后, C,N及S的烧除率分别为94.46%,95.35%及95.97%, 积碳基本脱除, 但仍有少量C,N等杂质存在, 活性金属流失不明显; 再生后, 催化剂物相基本得到恢复, 活性金属分散均匀, 团聚不严重; 再生后, 催化剂表面元素变化不大, 出现少量杂质元素, 金属元素W和Ni主要以Ni-W-S活性相存在。
3) 在相同加氢工艺条件下, 新鲜催化剂的脱硫、脱氮率为99.63%和99.76%, 而再生催化剂分别为99.45%和99.24%。再生催化剂对反应物DBT和QI的转化率以及产物选择性良好。
| [1] |
陈晓珍, 崔波, 石文平, 等. 催化剂的失活原因分析[J]. 工业催化, 2001, 9(5): 9-16. DOI:10.3969/j.issn.1008-1143.2001.05.002 |
| [2] |
梁相程, 王继锋, 连丕勇, 等. 几种加氢预处理催化剂的失活原因探讨[J]. 石油化工高等学校学报, 2001, 14(2): 50-58. DOI:10.3969/j.issn.1006-396X.2001.02.011 |
| [3] |
刘颖, 韩崇仁, 方维平, 等. 催化剂积碳失活宏观反应动力学研究[J]. 催化学报, 2004, 25(2): 107-109. DOI:10.3321/j.issn:0253-9837.2004.02.007 |
| [4] |
郑宇印, 刘百军. 加氢精制催化剂研究进展[J]. 工业催化, 2003, 11(7): 1-6. DOI:10.3969/j.issn.1008-1143.2003.07.001 |
| [5] |
孙万付, 马波, 张喜文, 等. 工业Mo-Ni/USY-Al2O3失活催化剂的再生行为[J]. 催化学报, 2000, 21(2): 157-160. |
| [6] |
张海永.低温煤焦油加氢处理用NiW/γ-Al2O3催化剂的研究[D].北京: 中国矿业大学, 2012. http://cdmd.cnki.com.cn/Article/CDMD-11413-1013294233.htm
|
| [7] |
张喜文, 凌凤香, 孙万付, 等. 一种失活加氢精制催化剂再生行为的研究[J]. 当代化工, 2005, 34(5): 52-56. |
| [8] |
REINHOUDT H R, CREZEE E, LANGEVEI D, et al. Charaeterization of the aetive phase in NiW/γ-Al2O3 catalysts in various stages of sulfidation with FTIR(NO) and XPS[J]. J Catal, 2000, 196(2): 315-329. DOI:10.1006/jcat.2000.3042 |
| [9] |
孙万付, 马波, 索继栓, 等. 加氢裂化催化剂积碳行为的研究[J]. 催化学报, 2000, 21(3): 269-272. DOI:10.3321/j.issn:0253-9837.2000.03.019 |
| [10] |
SIOKOU A, LEFTHERIOTIS G, PAPAEFTHIMIOU S, et al. The effect of the tungsten and molybdenum oxidationstates on the thermal coloration of amorphous WO3 and MoO3 films[J]. Surface Science, 482-485(part1): 294-299. |
| [11] |
YANG S H, SATTERFIELD C N. Catalytic hydrodenitrogenation of quinoline in a trickle-bedreactor. Effect of hydrogen sulfide[J]. Industrial & Engineering Chemistry Process and Development, 1984, 23(1): 20-25. |
| [12] |
张世万.煤焦油催化加氢轻质化及催化剂的研究[D].上海: 华东理工大学, 2011. http://cdmd.cnki.com.cn/Article/CDMD-10251-1012309953.htm
|
| [13] |
BUTAILLE F. Alkyldibenzothiophenes hydrodesulfurization-promoter effect, reactivity and reaction mechanism[J]. J Cat, 2000(191): 409-422. |
| [14] |
刘坤, 刘晨光, 李望良. Mo-Ni-P柴油加氢精制催化剂的研制[J]. 石油学报(石油加工), 2001, 17(5): 82-88. |
| [15] |
MIKI Y, SUGIMOTO Y. Hydrodenitrogenation of isoquinoline[J]. Appl Cat A, 1999(180): 133-140. |