 2019, Vol. 47
2019, Vol. 47文章信息
- 常增花, 王建涛, 李文进, 武兆辉, 卢世刚
- CHANG Zeng-hua, WANG Jian-tao, LI Wen-jin, WU Zhao-hui, LU Shi-gang
- 锂离子电池硅基负极界面反应的研究进展
- Research progress on interface reaction of silicon-based anode for lithium-ion battery
- 材料工程, 2019, 47(2): 11-25
- Journal of Materials Engineering, 2019, 47(2): 11-25.
- http://dx.doi.org/10.11868/j.issn.1001-4381.2018.000450
-
文章历史
- 收稿日期: 2018-04-22
- 修订日期: 2018-06-28




2. 国联汽车动力电池研究院有限责任公司, 北京 100088
2. China Automotive Battery Research Institute Co., Ltd., Beijing 100088, China
环境污染和能源危机使得绿色能源技术得到迅猛发展,锂离子电池以能量密度高、工作电压高、循环寿命长、环境污染小等优势广泛应用于手机、数码相机、笔记本电脑等各类小型便携式装置中,并有进一步作为动力和储能电源取代传统镍镉和铅酸等电池的趋势,已成为当今世界极具发展潜力的新型绿色高能化学电源。近年来人们开始广泛研发锂离子电池在电动汽车方面的应用。当前纯电动汽车大规模产业化所面临的第一大障碍,就是续航里程的问题,其续航里程是由动力电池系统所能够释放出来的电能决定的,因此动力系统的能量密度就成了制约电动车续航里程的决定性因素。对于锂离子电池而言,其理论能量密度由正负极材料比容量和工作电压决定。提高锂离子电池的能量密度主要有两个途径:一是提高电池工作电压,二是提高正负极材料的比容量。
而欲将锂离子电池动力汽车达到目前具有实际推广意义的300km以上续航里程,其电池的能量密度需要达到200~250Wh/kg,对于负极而言,传统石墨材料受理论容量的限制,难以满足该能量密度要求。然而,Si负极材料比传统石墨材料的质量比容量提高了十余倍,因此Si基负极材料的研究受到广泛关注[1-3]。然而目前Si负极材料还面临着几个重要的问题,限制了Si负极材料在锂离子电池中的商业化应用。这些问题包括:(1)电子导电性和离子导电性差,不利于材料电化学性能的发挥[4-6];(2)嵌/脱锂过程中伴随着巨大的体积变化,引起Si颗粒产生裂纹粉化,导致容量迅速衰减,从而严重地影响了Si负极的循环性能;(3)Si负极与电解液的界面不稳定[7],Si负极材料表面形成的钝化膜不能适应Si负极材料在脱嵌锂过程中的巨大体积变化[8-9]而破裂,新鲜的Si表面暴露在电解液中,导致钝化膜持续生成、活性锂不断消耗,最终造成容量损失[10]。深入了解Si负极的界面反应,有利于针对Si负极界面问题提出相应的改善措施,从而提高Si负极的电化学性能。Si负极界面化学的演化机制包括3个方面:Li-Si合金化过程、表面氧化硅的反应和钝化膜的形成[11]。Si电极容量损失也主要来源于这3个方面,首次循环以及后续循环过程中电解液在Si表面还原形成钝化膜,导致Li+损失;初始容量损失的另一个可能的原因是SiO2的还原;Li在活性物质Si中的不可逆嵌入也会导致部分容量损失[12]。
本工作综述了锂离子电池硅基负极中Li-Si合金化,表面氧化硅反应和钝化膜形成机制,并分析了影响Li-Si合金化的因素以及表面氧化硅反应和钝化膜形成对硅基负极电化学性能的影响。
1 硅基负极的合金化过程 1.1 硅的储锂机制Si负极为合金化储锂机制,Si与Li的合金化反应可以描述为:
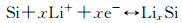
|
(1) |
根据是否有相变发生,该反应可以分为固溶反应(solid-solution (insertion) reaction)和加成反应(addition reaction)两种类型[13-14]。固溶反应中,Li嵌入Si中没有相转变或者结构变化,是一种局部异构反应;加成反应中,Li嵌入Si中存在相转变或者结构变化。无定形Si与锂的合金化属于固溶反应,而晶体Si与锂的合金化属于加成反应。
恒定温度和压强下,吉布斯相律可用下式表示:
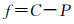
|
(2) |
式中:C为独立组元数; P为相数; f为封闭体系在平衡态的自由度。
在Li-Si两相固溶反应中,C=2,P=1,则f=1,反应电位随着锂浓度的变化而变化,电压曲线表现为斜线(sloping shape);在Li-Si加成反应中,C=2,P=2,则f=0,平衡态下反应电位与成分无关,电压曲线上出现平台,对应着两相平衡区。
Li-Si反应的电位可以通过Nernst方程求出:
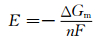
|
(3) |
式中:ΔGm为LixSi的形成自由能; n为参与反应的电子数; F为法拉第常数。
在高温下(415℃),Si与Li发生电化学合金化反应时遵循Li-Si相图,经历了多相转变,分别形成了Li2Si7, Li7S3, Li13Si4, Li22Si5四个相,其充放电曲线分别对应0.332, 0.288, 0.158和0.044的电压平台[13, 15-17]。而常温下的嵌脱锂反应偏离了平衡态,Si的首次放电(嵌锂)曲线在约0.1V出现一个较低的电压平台(如图 1所示)[17],这是由于c-Si转化为部分嵌锂相a-LixSi,出现c-Si和a-LixSi两相共存,该两相区一直持续到50mV,电压降到50mV以下,a-LixSi突然转化为c-Li3.75Si,这种结晶化可能对应着微分电容曲线上< 50mV的小峰(如图 2(a)中的1峰)[2, 18]。此外,后续循环过程的合金化曲线没有电压平台,呈现圆弧状(round-shaped)。脱锂过程中,出现另一个两相区,c-Li3.75Si又转化为a-LixSi,进而转化为a-Si(如图 2(a)中的2峰)。再次嵌锂过程中,a-Si嵌锂依然保持无定形态的单向区,对应着容量微分曲线上的一个宽峰(如图 2(a)中的3峰)。如果电压降到50mV以下,a-LixSi又会转化为c-Li3.75Si。Si的充放电过程如反应(4),(5),(6)和(7)所示[19-20],式中c表示结晶态,a表示无定形态。


放电(合金化/嵌锂)过程:
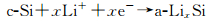
|
(4) |
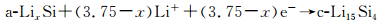
|
(5) |
充电(去合金化/脱锂)过程:
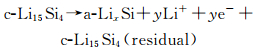
|
(6) |
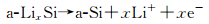
|
(7) |
首次嵌锂过程中,晶体Si转化为无定形相Li-Si而不是平衡态的晶相Li-Si。一般认为这是由于亚稳态无定形相Li-Si的自由能比平衡态的晶相低[21]。但是随后的循环过程中,嵌锂(无定形Si转化为无定形的Li-Si)主要发生在0~0.3V,这说明无定形Li-Si与晶相Li-Si的平衡电势非常接近,具有相近的自由能。因此Zhang等[13]推测大的成核能量势垒可能是无定形Li-Si代替晶相Li-Si的原因,并且给出了详细解释[13]。如图 3,平衡态无定形Li-Si相(AM,实线)的自由能高于Si和LixSi两相混合物(点线a-b),因此在平衡相图上预测了Si+LixSi两相区。Si,LixSi和AM相平衡自由能曲线(实线)指的是无穷大的尺寸。也就是说,颗粒尺寸足够大,与相的体积能量相比,界面/表面能可以忽略不计。但是在实际的合金体系中,新相在母相中的形成开始于小尺寸的晶核和晶核的长大,成核会产生新的相界面,因此需要克服能量势垒G*(图 4)。界面能(正)随r2增加,而体积能(负)随r3减小(r为球形核的半径),而体系总的自由能(G)通常在成核初期为正。晶核在达到临界尺寸R*(对应自由能最大值G*)之前处于热力学不稳定状态,高的界面能会导致大的临界晶核尺寸R*。在母相Si中形成稳定的LixSi晶核(LixSin,虚线)的实际总能量远高于该相的平衡自由能(LixSi,实线)。同样,稳定的无定形Li-Si核(AMn,虚线)的总形成能也要高于平衡值(AM,实线)。由于无定形相的无序结构和低堆积密度,其形成能和界面能比晶相低,所以与形成一个晶相核LixSin相比,形成一个稳定的无定形Li-Si核(AMn)从热力学讲是有利的。因此,Li嵌入Si是通过由晶体Si到无定形Li-Si(灰色实线a-c)的两相反应,而不是由Si到晶相LixSi(虚线a-b)的平衡反应。与晶相LixSi的平衡电位(a-b斜线)相比,无定形核(AMn)相对较低的反应电位(a-c斜线),可能来源于无定形相低的自由能和晶相高的界面能。

|
图 3 晶体Si、锂化相LixSi和非晶相AM (实线)平衡吉布斯自由 能曲线的示意图以及对应的小尺寸核的Sin,LixSin和AMn(虚线)总自由能曲线[13] Fig. 3 Schematic illustration of equilibrium Gibbs free-energy curves for crystalline Si, lithiated phase LixSi and amorphous phase AM at infinite sizes (solid lines) and total free-energy curves for corresponding nuclei of small sizes: Sin, LixSin and AMn (dash lines)(The total free energy of a stable nucleus is higher (less negative) than the equilibrium Gibbs free energy due to the additional interface energy)[13] |

如果锂化相是晶相LixSi(如Li15Si4),那么在脱锂过程中也会发生同样的情况。Li15Si4相按照b-d线生长为无定形的Li-Si核,而不是按照平衡线b-a形成晶相Li-Si或Si。如果在上一次合金化过程中没有形成结晶相,那么Li从无定形LixSi相脱出是一个没有相变的固溶反应(如果粒子足够大,则遵循AM自由能曲线),因而表现为倾斜的电压曲线。
1.2 颗粒尺寸对Li-Si合金化的影响从早期对硅负极材料的研究可以看出,Li-Si合金化过程(主要是晶相Li15Si4的形成)强烈依赖于硅颗粒尺寸。从李泓等[22]在微米硅(过250目筛)和纳米硅(78nm)中得到的充放电曲线可以看出,微米硅在0.45V左右有一个电压平台,该平台对应着c-Li15Si4向a-LixSi转化的两相区,而纳米硅脱锂过程表现出一个倾斜的电压曲线,说明该过程中只有a-LixSi到a-Si的转化过程,在嵌锂过程中可能没有c-Li15Si4生成。
Li等[23]采用原位XRD发现在大尺寸晶体硅(325 mesh size)中Li15Si4相在嵌锂电位约为60mV时会发生突然结晶。Zhang等[13]从能量的角度给出了相应的解释,Li嵌入无定形LixSi相,使Li的浓度增加到Li15Si4的组成,该嵌锂相的总能量在线AMn和AM之间(图 3)。在嵌锂末期,无定形LixSi相的组成与晶相Li15Si4非常相近。晶相(b点)的平衡自由能比无定形相低,但由于成核所需的界面能较高,从无定形的LixSi到晶态Li15Si4的相变仍然是不可行的。然而,如果无定形颗粒的尺寸足够大,以至于结晶过程中表面/界面能的增加与体积能的降低相比较小,有利于无形核相变。相变可以通过非扩散过程进行,而不需要改变成分。在这种情况下,一旦发生相变,整个无定形的粒子将会通过近程原子重排突然转变成晶相。当颗粒尺寸较小时,小颗粒两相反应的总能量将由界面能而不是体积能量支配(图 4)。因此,由于第二相形核导致的能量增加,会因颗粒尺寸减小而升高。当颗粒尺寸小于临界值时,这些颗粒中新相的成核是热力学不稳定的,而粒子则倾向在整个范围内与Li固溶。换句话说,大颗粒的加成反应和小颗粒的固溶反应是热力学有利的,在纳米颗粒中观察到的倾斜电压曲线可能是Li与Si固溶的结果。
而Gao等[24]通过检测放电过程中静态漏电流给出小颗粒硅材料能够抑制循环过程中相转变的另一种解释,由于小尺寸的硅材料在0.05V左右与碳酸酯类电解液之间有快速的副反应发生,所以即使在嵌锂过程中有c-Li15Si4形成,在放电结束后也会消失,如图 5所示。

在合金化过程中Li-Si相转变以及在去合金化过程中形成的相强烈依赖于嵌锂截止电压和嵌锂倍率。近几年,采用一系列原位和非原位的方法,包括原位NMR、原位XRD、原位TEM和非原位对分布函数分析等研究了Li-Si体系。这些研究表明在首次合金化过程中,c-Si相转化为a-LixS相,电位较低时(< 50mV vs. Li),a-LixS相会转化为完全嵌锂相c-Li3.75Si[3, 23, 25-30]和过度嵌锂相如c-Li3.75+δSi[23, 31]或Li4.4Si/Li4.2Si[25, 32]。如果完全合金化到0V,去合金化曲线在~0.4V会出现一个平台(图 6(a)[33])。这是因为在合金化过程中形成的c-Li3.75Si,在脱锂时又通过加成反应部分转化为无定形态a-LizSi(z≈2)。如果合金化的截止电位在0.05V以上,合金化的最终产物为无定型相a-LixSi,去合金化过程是由无定形相LixSi生成无定形Si的固溶反应,因此出现倾斜的电压曲线,没有明显的电压平台(图 6(b)[33])。

英国剑桥大学Ogata等[18]采用原位核磁共振波谱法研究了硅纳米线(SiNWs)的嵌锂和脱锂行为,在第一周及后续循环过程中,SiNWs的嵌/脱锂行为如表 1所示。若用C/25的电流以恒流方式在0~2V的电压范围内进行嵌锂,在第二周及后续的循环中,Si的锂化过程可以分为4步:第一阶段(Si#d2),放电电压为300~250mV时,a-Si点阵缓慢嵌锂形成Li~2.0Si,Li~2.0Si相仍包含延伸的Si网和较大的Si—Si簇;第二阶段(Si#d3):电压为100mV时,Si—Si键的进一步断裂,形成较小硅簇,最终形成孤立的Si原子,此阶段形成Li~3.5Si相;第三阶段(Si#d4):电压降到50mV,部分小硅簇(a-LixSi(x=2.0~3.5))开始形成少量孤立的Si4-负离子和a-LixSi(x=3.5~3.75);残留的a-LixSi相中存在少量的孤立的Si4-负离子,表明c-Li3.75Si相在SiNW富含孤立Si4-负离子的区域非均匀形核,剩下富含小Si—Si簇的a-LixSi相作为c-Li3.75Si成核生长的副产物。c-Li3.75Si容易在富含孤立Si4-负离子的a-LixSi区域形核生长,因为这个过程不需要Si—Si的断裂,c-Li3.75Si在a-Li3.5-3.75Si区域的非均匀成核导致出现贫锂区域也就是富含小Si—Si簇的a-LixSi相。第四阶段(Si#d5):电压降到30mV以下时,出现过度嵌锂相c-Li3.75+δSi。非原位XRD证实电压低于30mV时,c-Li3.75Si依然存在,没有观察到明显的新相(例如Li4.2Si,Li4.4Si),暂时认为这个扩展到30mV的过程相为过度嵌锂相c-Li3.75+δSi(δ=0.2~0.3),在结构上与c-Li3.75Si相关。小硅簇(a-LixSi(x=2.0~3.5))和Si4-/a-LixSi(x=3.5~3.75)依然存在,并出现c-Li3.75Si和过度嵌锂相c-Li3.75+δSi,a-Li3.5~3.75Si转化为c-Li3.75Si伴随着c-Li3.75Si转化为c-Li3.75+δSi,与下面讲到的慢循环相比,Si4-/a-LixSi(x=3.5~3.75)的含量较少,a-Li3.5~3.75Si转化为c-Li3.75Si的动力学阻力比c-Li3.75Si转化为c-Li3.75+δSi的阻力大,因此该过程中观察到了这两个转化步骤。如果嵌锂截止电压为50mV,此时电极材料中含有小硅簇和小硅簇与Si4-负离子的混合物,然后开始充电,进行脱锂过程,270mV时,a-Li3.5Si转化为a-Li2.0Si,500mV时,a-Li2.0Si转化为a-Si;如果放电到0V再次充电开始脱锂过程,30~80mV时,c-Li3.75+δSi转化为c-Li3.75Si,并有少量小硅簇生成,270mV时,小硅簇a-Li3.5Si转化为大硅簇a-Li2.0Si,430mV时,少量c-Li3.75Si转化为a-Li1.1Si,500mV时,a-Li2.0Si转化为a-Si。
| Lithiation process | De-lithiation process |
| 1st discharge | Si#c1 |
| Si#d1 | Li3.75+δSi→c-Li3.75Si+ c-Li3.75-δSi |
| c-Si→a-LixSi | Si#c2 |
| →c-Li3.75Si | a-Li3.5Si →a-Li2.0Si |
| →c-Li3.75+δSi | Si#c3 |
| 2nd discharge | c-Li3.75Si→a-Li1.1Si |
| Si#d2 | Si#c4 |
| a-Si→a-Li2.0Si | a-Li2.0Si→a-Si |
| Si#d3 | |
| a-Li2.0Si→a-Li3.5Si | |
| Si#d4 | |
| a-Li3.75Si →c-Li3.75Si | |
| Si#d5 | |
| c-Li3.75Si →c-Li3.75+δSi |
以恒流(电流C/25)放电至不同的截止电压:第一周放电至0V,确保c-Si完全转化成为a-Si;在后续循环过程中,以10mV为一个梯度,从150mV依次降低至0V;然后提升至180mV,以10mV为梯度再依次降低。当放电截止电压高于110mV时:嵌锂主要发生在Si#d2过程,脱锂发生在Si#c4过程中,从a-Li~2.0Si中脱锂形成a-Si;当放电截止电压低于110mV时,嵌锂开始出现Si#d3过程,因此在脱锂过程中出现了Si#c2过程,孤立的硅原子生长形成小硅簇。当放电截止电压低于50mV时,脱锂过程中在430mV出现了一个尖锐峰(Si#c3),对应于晶态结构的c-Li3.75Si转化成无定型结构的a-Li1.1Si;如果放电到0V后,再次充电开始脱锂,电压在50~150mV范围内时,c-Li3.75+δSi转化成c-Li3.75Si的结晶相和有缺陷c-Li3.75-δSi结晶相(Si#c1);进一步充电,电压达到430mV时,c-Li3.75Si/c-Li3.75-δSi转化为a-Li~1.1Si(Si#c3)。值得一提的是,及时完全放电至0V,在充电过程中仍旧可以看到Si#c2过程,这是由于在C/25的电流下所有的硅纳米线很难完全转化为c-Li3.75Si晶体相,然后通过Si#c2和Si#c4过程转化成a-Si。
总之,在脱锂过程中形成的相强烈地依赖于嵌锂倍率和截止电压,过低的嵌锂截止电压,会出现无定形相a-Li3.75Si向结晶相c-Li3.75Si的转化,两相区的存在导致较大的体积变化,伴随着高的内部应力,导致颗粒开裂、电接触恶化和容量衰减。因此,在硅基负极使用过程中,合理地控制嵌锂电压下限,使电极在整个循环过程中都保持无定形态,可获得较好的循环性能。
2 硅基负极表面氧化硅的反应Si表面有一层原生氧化硅(SiOx,具体成分与反应条件有关),尤其是SiO2(其他氧化硅成分含量较少)[34],SiO2的实际反应途径和最终产物及其对Si电极电化学性能的影响目前还不是非常清晰,依然存在争议,许多文献中的报道也相互矛盾,尚无一致的看法[35-36]。
在电极循环过程中,SiO2可能的反应方式有两种:(1)在电极嵌锂过程中发生电化学还原;(2)电解液还原产物进一步与SiO2发生化学反应。
2.1 SiO2的电化学反应在Li嵌入Si电极时,Si表面的SiO2可能通过两种反应途径被还原,反应(8)和(9) [12, 34, 37-38]。然而,反应能否进行,许多文献的报道并不一致。
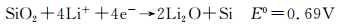
|
(8) |
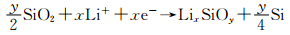
|
(9) |
Fultz等[39]认为Li还原SiO2是热力学有利的,因此导致较高的首次不可逆容量,但没有给出SiO2还原的证据。相反,Saint等[40]由热力学计算获得反应(8)的ΔG0为负值,相应的E0为正值,并认为电极电位必须低于平衡电位约1V时才能引发反应(8),所以认为在首次嵌锂过程中SiO2通过反应(8)被还原的可能性非常小,动力学上几乎是不可能的。而Lee等[41]根据EIS测得首次嵌锂过程中总阻抗先减小后增大的结果,认为在首次嵌锂的初始阶段Si电极的原生表面层(SiO2和Si—OH)被破坏导致阻抗减小,但是表面层的破坏也可能是化学反应的结果。
后来,在不同的研究中,采用XPS和NMR观察到循环过程中SiO2的还原导致新相的生成,被认为是Li4SiO4[12, 37]或Li2Si2O5[42],相应的电化学反应为(10)和(11)。
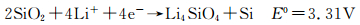
|
(10) |
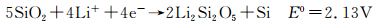
|
(11) |
虽然已经观察到Si电极表面SiO2膜的嵌锂行为,但是关于该反应的可逆性依然存在分歧。
陈立泉等[37]研究了水热合成法制备的硬碳-纳米SiO2复合材料(HC-SiO2)的电化学还原过程,NMR和XPS的测试结果以及高的储锂容量表明纳米SiO2在首次放电过程中可以通过电化学还原反应(8)和(10)生成Li2O,Li4SiO4和Si,首次循环的还原产物Si可通过反应(12)实现可逆嵌锂,但反应(8)和(10)不可逆。
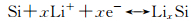
|
(12) |
复旦大学傅正文等[42]将SiO2薄膜沉积到不锈钢基体上作为电极,在0.01~3.0V之间获得416~510mAh·g-1的可逆容量。根据HR-TEM、SAED和XPS的检测结果,认为SiO2薄膜可发生电化学反应(11)和(12)。
Philippe等[12]采用Hard X-ray PES在Si/C/CMC复合电极首次循环后的表面深层检测到Li2O和Li4SiO4,认为(8)和(9)两种反应可能同时存在。由于采用的是高能光子2300eV(甚至更高6900eV),认为Li2O和Li4SiO4在SEI膜和核心颗粒Si/LixSi之间。还发现Li2O的形成不只是发生在开始嵌锂形成Li-Si合金时,直到嵌锂结束依然有Li2O形成。此外,Li2O的形成可能是可逆的,嵌锂过程中形成的Li2O,在脱锂过程中逐渐消失。当然,Li2O的消失也可能是HF的影响,Li2O很容易与HF反应生成LiF[7]。
Radvanyi等[34]采用XPS/AES比较研究了纳米Si电极表面SiO2在充放电过程中的行为。如图 7,循环前的电极A,检测到Si颗粒表面存在SiO2层;嵌锂容量为360mAh·g-1的电极B表面SiO2的信号消失,出现LixSiOy相,认为是SiO2发生电化学还原的结果,但是不能确定LixSiOy的具体成分,可能包括Li4SiO4,Li2Si2O5或Si的低氧化物,伴随着LixSiOy的生成,电极表面也出现了另一相,可能为Li2O;完全嵌锂电极C,Si相消失,检测到LixSi和Li2O。以上结果表明在首次嵌锂过程中,SiO2被还原为Si或LixSiOy(根据反应(8)和(9)),而且在进一步嵌锂过程中,SiO2在持续还原,这是因为随着嵌锂的进行,Li2O相应的信号在增加。完全脱锂电极D,从其XPS分析可知,存在LixSiOy,但Li2O消失,可能是发生了反应(13):
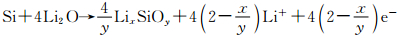
|
(13) |

|
图 7 硅电极表面的XPS谱图 (a)原始电极; (b)放电360mAh·g-1;(c)放电3800mAh·g-1;(d)一次完整的充放电循环后 Fig. 7 XPS spectra (Si2p, C1s, O1s, and Li1s core peaks) obtained at the surface of the Si electrodes (a)pristine electrode; (b)after a capacity of 360mAh·g-1; (c)after a capacity of 3800mAh·g-1; (d)after a complete electrochemical cycle |
与Philippe等[12]的报道相反,脱锂后(电压达到1.2V)没有发现SiO2,在这种情况下,反应(8)和(9)是不可逆的。循环5次后的XPS结果(图 8)表明反应(13)是可逆的,由此看来初始SiO2层的彻底还原对Si颗粒的表面化学性质有强烈影响。Philippe等[12]报道首次循环后Si颗粒表面还存留一些SiO2,SiO2与电解液中LiPF6反应转化为SiOwFz,Philippe等认为颗粒表面氟化物的形成使Si和黏结剂之间的相互作用恶化,导致电极性能的迅速衰减。而Radvanyi等[34]在循环5周后的电极表面未观察到氟化组分,因此得出结论:(1)如Philippe等所述SiOwFz的形成反应涉及SiO2;(2)貌似可以通过不改变电解液中锂盐的方式来避免氟化组分的形成。总之,Radvanyi等采用XPS表征了Si颗粒表面SiO2的演变过程。首次嵌锂时,SiO2经电化学还原形成LixSiOy和Li2O;脱锂时Li2O与Si反应形成LixSiOy,该反应在后续的循环过程中是可逆的。SiO2被还原后在后续循环中没有再出现,原生氧化层的彻底消失可能对电极的循环特性有较大影响,所以SiO2完全被还原的条件如初始厚度、Si形态、循环条件有待进一步研究。

|
图 8 第5周循环过程中Si电极表面的XPS分析 (a)1.2V脱锂后; (b)0.005V嵌锂后 Fig. 8 XPS analysis obtained at the surface of Si electrode during the fifth electrochemical cycle (a)after delithiation at 1.2V;(b)after lithiation at 0.005V |
Schroder等[43]发现含有氧化硅层的Si电极和刻蚀掉氧化层的Si电极电化学反应的主要不同在于图 9(a)比图 9(b)多了一个还原峰,该还原峰出现在0.47~0.59V之间,Schroder等将其归为至少部分氧化硅的电化学还原。

|
图 9 (100) 晶片工作电极在1mol·L-1 LiPF6-EC/DEC (1:1, 质量比)电解液中的典型循环伏安曲线 (a)原生层; (b)RIE刻蚀表面 Fig. 9 Representative cyclic voltammograms using 1mol·L-1 LiPF6 in EC/DEC (1:1, mass ratio) and (100)silicon wafer working electrodes (a)with a native oxide; (b)with a RIE etched surface. Both electrodes were cycled from OCV (about 3.0V) to 0.01V Li/Li+ at 10mV·s-1 scan rate for 5 cycles |
根据化学反应热力学,可通过标准电极电势E0来判断反应的可能性,其依据也就是Gibbs函数变ΔG0。由标准电极电势E0可知,反应(8),(10)和(11)在负极的工作电位范围内(1~2.01V)正向进行是可能的,但是产物Li2Si2O5和Li4SiO4在0.01~1V之间是相当稳定的,尽管这些硅酸盐在2V(Li+/Li)以上可以脱锂,但锂离子电池负极工作电位达不到2V,而Li2O脱锂则是可能的。
虽然从热力学上讲,SiO2的还原是可能的,然而在正常的循环条件下SiO2的还原可能是非常缓慢的[40],要使反应能够进行可能需要极端的循环条件,例如较高的过电位和较长的时间。也就是说事实上SiO2能否被电化学还原,依赖于反应的条件,但至少可以说明,在一定的条件下,SiO2的电化学还原是可以实现的,可能(8)和(9)两种反应同时存在。尤其是在较低的电压下,可能还原反应的速度还是非常快的[35]。此外,以SiO2薄膜材料作为电极获得了放电容量[42],也说明SiO2是可以发生电化学反应的。至于还原产物的可逆性,根据热力学分析及文献报道结果Li2O的生成可能是可逆的,而硅酸盐类产物在负极工作电位范围内(0.01~1V)很稳定。与SiO2相比,其还原产物的电子导电性及Li+离子导电性均增加。
2.2 SiO2的化学反应由于LiPF6不稳定与微量的H2O反应生成HF,HF与SiO2在Si电极表面反应,侵蚀SiO2。Mitra等[44]提出了SiO2溶于HF溶液的反应机理,认为该过程包括H+对桥氧原子Si—O亲电子攻击和HF2-对邻近Si原子的亲核攻击两个基元反应,如图 10。而Philippe等[7]在电极材料颗粒的表面发现了SiOxFy,表明HF与SiO2在电池里的反应比以往提出的直接生成不稳定的SiF4的催化反应路径(反应(14))复杂得多。同时,Li2O与HF反应,产物H2O可进一步生成HF。在循环过程中,Li2O不断消耗,氟化物不断生成。
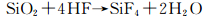
|
(14) |
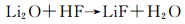
|
(15) |

此外,通过对Si2p光电子能谱的分析,给出了在循环或储存过程中硅负极与电解液界面化合物的形成过程,如图 11。初始状态下Si/C/CMC电极中Si颗粒表面存在少量SiO2;与电解液接触放置96d后,氧化物层(SiO2)几乎消失,出现氟化物SiOxFy,覆盖Si表面;首次部分放电后,SEI膜形成,Li嵌入Si体相中,在SiO2层中生成Li4SiO4;首次放电后与电解液接触182d后,没有氟化合物生成,体现了SEI膜的保护作用,SEI膜一旦生成,就像一个屏障,可以阻止HF酸到达氧化物层;但是,在循环过程中(有电压存在的情况下),这个屏障允许HF酸迁移通过,与SiO2反应,在SEI膜与剩余的SiO2层之间生成氟化合物SiOxFy;电压终止后,没有发现进一步的反应,SEI膜再次起到保护作用。

|
图 11 循环或储存过程中Si颗粒表面的XPS图谱及Si颗粒与电解液界面形成化合物示意图[7] (a)Si2p谱(内置PES, 1486.6eV);(b)循环或储存过程中Si颗粒与电解液界面形成化合物示意图(右图);①Si/C/CMC未循环电极;②在电解液里浸泡96天的电极;③首次放电500mAh/g的电极;④第100次放电到0.12V(预循环)的电极(左图) Fig. 11 XPS spectra of Si particle surfaces and schematic of compounds formed at the interface of Si particles and electrolyte during cycling of storage[7] (a)Si2p spectra (in-house PES, 1486.6eV); (b)schematic view of the compounds formed on the silicon particle surfaces during cycling and/or during storage with the electrolyte (to the right); ①Si/C/CMC pristine electrode; ②a pristine electrode in contact 96 days with electrolyte; ③after a first discharge until 500mAh/g of Si; ④after the 100th discharge at 0.12V (with precycling) (to the left) |
总之,通过对活性物质表面的分析,在循环过程中或原始电极与电解液接触放置时,在Si颗粒和SEI膜之间的界面均会生成氟硅化合物(SiOxFy,y≤3);Li2O相只在嵌锂状态下出现,在循环过程中逐渐消失。这两个现象均体现了LiPF6分解生成的HF对电极表面的影响。
2.3 电解液在SiO2表面的还原美国阿贡国家实验室Hubaud等[45]采用耗散型电化学石英晶体微天平(EQCM-D)分析了Si薄膜(100nm)和Si-SiO2(50~50nm)薄膜电极上SEI膜的形成,在高于嵌锂电位时,在这两种电极上均检测到负向频移值,即质量均增加,说明在有电位存在的情况下,两种电极表面均与电解液发生了反应。与Si电极相比,Si-SiO2电极的频移值较小,且增重峰和电流峰出现的时间延后,可能是由于SiO2的绝缘性特性和电容造成的。
2.4 SiO2的影响 2.4.1 SiO2对钝化膜的影响Schroder等[43]采用XPS分析了含有氧化硅层的Si电极和刻蚀掉氧化层的Si电极在1mol·L-1 LiPF6 EC/DEC (1:1,质量比)电解液中形成的SEI膜的组成及结构。含有氧化硅层的Si电极表面形成的SEI膜含有较多的有机组分,刻蚀后的Si电极上形成的SEI膜含有大量的无机组分如LiF和PxOyFz,以及少量的LiOx。LiOx的含量虽少,但意义重大,在含有氧化硅层的Si电极表面形成的SEI膜中并未发现LiOx。Schroder等建议可用两种Si电极竞争成膜反应的动力学及电子和化学性质的不同来解释SEI膜组分的差别。还通过TOF-SIMS法研究了SiOx对SEI膜厚度的影响,结果表明与含有氧化硅层的Si电极相比,采用相同的电化学方法在刻蚀Si电极上形成的SEI膜较厚。
此外,Mitra等[44]认为电解液中的HF会侵蚀SiO2导致Si表面形成的SEI膜不稳定,而Dirican等[46]则认为Si表面包覆SiO2可避免活性物质Si与电解液的直接接触从而可形成较稳定的SEI膜。崔屹等[47]为了解SiO2的影响,对比研究了硅纳米线(SiNWs)电极和HF刻蚀后的SiNWs电极,刻蚀后的SiNWs在电解液中浸渍几分钟,用DMC冲洗,经XPS分析发现表面含有电解液还原产物,包括碳氢化合物、PEO型低聚醚、烷基碳酸锂、LiF和LiPFxOy,此外还有含氟有机物(相同条件下,刻蚀后的硅晶片中未发现,笔者认为可能是由于SiNWs大的表面积导致与电解液高的反应活性)。这是由于HF刻蚀的SiNWs表面有高活性的Si—H,会与电解液形成不同的表面膜,含有相当一部分含氟有机化合物,而未刻蚀的SiNWs表面的SiO2处于钝化状态,在其表面只能生成普通的电化学还原产物。
2.4.2 SiO2对硅电极电化学性能的影响目前普遍认为Si电极表面SiO2的存在会导致低的容量和低的库仑效率,但对电极的循环稳定性是有利的。Xun等[48]通过改变HF刻蚀时间来控制Si纳米颗粒表面氧化层厚度,并采用TEM、XPS和TGA等方法研究了Si纳米颗粒表面氧化层厚度对电池性能的影响,发现表面氧化层对Si纳米颗粒电极的首次循环性能存在不利的影响,导致可逆容量降低,通过减小氧化硅层的厚度可以提高Si纳米颗粒电极的可逆容量。由此可知,氧化层厚度不同可能是导致市售Si纳米颗粒性能不同的一个重要原因。崔屹等[47]发现HF刻蚀后的SiNWs由于去除了表面SiO2,首次循环效率从原始的76%提高到89%,但是循环性能变差,如图 12所示。由于未刻蚀的SiNWs电极充电到0.5V时,SiO2已消失,所以决定电极长期循环性能的因素可能不是SiO2的存在,而是SiNWs上形成的表面膜。

|
图 12 HF刻蚀和未刻蚀SiNWs在0.2C恒电流循环的容量变化 Fig. 12 Capacity vs. cycle number for galvanostatic cycling of HF-etched SiNWs and pristine SiNWs (un-etched) at the 0.2C rate |
Wu等[49]用纳米SiO2包覆Si纳米管以及Dirican等[46]在Si@C表面包覆纳米无定形SiO2层均使Si电极循环稳定性提高。认为初始嵌锂过程中,SiO2可能被还原为Li2O和Li4SiO4,有助于适应Li-Si合金化过程的体积膨胀,所以纳米SiO2包覆有利于保持Si电极在循环过程中结构的完整性,从而可提高电极的循环稳定性。McDowell等[36]研究了表面氧化层对SiNWs体积膨胀和循环性能的影响,认为表面氧化层会诱导静压应力,这种压应力具有导致Li-Si合金相中Li平衡浓度降低的热力学倾向,从而抑制Si在嵌锂过程中的体积膨胀;此外,不含氧化层的SiNWs在首次嵌锂过程中会发生更多的副反应(1.7~0.4V电压范围内充电曲线出现倾斜),但是具有更高的容量和更稳定的循环性能。
3 硅基负极表面钝化膜的形成 3.1 钝化膜的形成机理Goodenough等[50]分析了锂离子电池中电解液发生分解的根本原因,如图 13所示。电解液的电化学窗口为电解液最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能量间隔Eg,正负极的电化学势分别为μC和μA(也就是它们的费米能级)。当LUMO < μA,即电解液组分(溶剂或锂盐等)的LUMO低于负极的费米能级时,电子从负极注入电解液的LUMO,导致溶剂或锂盐被氧化(除非存在可以阻碍电子从电解液HOMO转移到正极的钝化膜);当HOMO > μC,即电解液组分(溶剂和锂盐等)的HOMO高于正极的费米能级,电子从电解液注入正极,导致溶剂或锂盐被氧化(除非存在可以阻碍电子从电解液HOMO转移到正极的钝化膜)。由于电解液的电化学窗口不够宽,一般在电池充放电过程中,电解液组分(包括溶剂和锂盐)会在电极表面发生还原或氧化,产生的不溶物将沉积在电极表面,形成固体电解质膜(SEI),有时也称表面钝化膜(surface passivating film)。SEI膜能够导通Li+,但是电子绝缘,因此能够阻碍电解液组分的进一步还原或氧化。

|
图 13 电解液的开路能量示意图(ΦA和ΦC分别为负极和正极的功函数,Eg是电解液的热力学稳定窗口,LUMO < μA和/或HOMO > μC形成SEI膜需要的动力学稳定条件)[13] Fig. 13 Schematic open-circuit energy diagram of an aqueous electrolyte(ΦA and ΦC are the anode and cathode work functions.Eg is the window of the electrolyte for thermodynamic stability.A μA > LUMO and/or a μC < HOMO requires a kinetic stability by the formation of an SEI layer)[13] |
热力学稳定性要求电极的电化学势μC和μA应在电解液的电化学窗口范围内,限制了电池的开路电压Voc(eVoc=μA-μC≤Eg),而电极/电解液界面钝化膜的存在可为更大的Voc提供动力学稳定性(如果eVoc-Eg不太大)。目前商用电解液不发生氧化还原反应的电化学窗口一般为1.2~3.7V vs Li+/Li。而锂离子电池工作电位范围一般为2~4.3V,其中石墨类负极工作电位范围在0~1.0V vs Li+/Li,正极工作电位范围一般在2.5~4.3V vs Li+/Li,因此在电极与电解液的界面不可避免地会生成SEI膜[50]。
3.2 钝化膜的形成电极在首次循环过程中,当电位降到电解液组分的还原电位时,电解液组分开始在电极表面还原,钝化膜开始形成。崔屹等[47]采用XPS,EIS和SEM等研究了Si纳米线(SiNWs)在1mol·L-1 LiPF6-EC/DEC (1:1, 体积比)电解液中表面钝化膜的形成过程及表面化学和形貌特性,主要特点总结如下:(1)钝化膜的形成过程依赖于电位的变化,首次放电至0.6V时钝化膜开始形成,钝化膜的大量形成发生在嵌锂电位,也就是说钝化膜除了在嵌锂电位以上形成,在嵌锂过程中,Si体积膨胀的同时,也伴随着钝化膜的形成。(2)钝化膜的形貌也依赖于电位的变化,在较低的放电电位下,钝化膜较厚,厚度已经超过了XPS的检测范围,约有40nm,含有大颗粒;随着充电电位的升高,钝化膜部分溶解并出现破裂现象。通过改变截止电压避免SEI膜形貌发生较大变化可以提高循环性能,因此认为SiNWs的循环性能强烈依赖于钝化膜。(3)Si负极表面的SEI膜成分与石墨负极基本一致,SEI膜的成分在其形成过程中也发生着相应的变化。
Yen等[51]研究了Si和C-coated Si电极在1mol·L-1 LiPF6-EC/EMC(1:2, 体积比)中形成的SEI膜,在两种电极上形成的共同成分有Li2CO3, LiF, ROCO2Li, ROLi和PEO,与石墨电极上形成的SEI膜成分一致,这些物质的形成反应在相关文献中已有报道。Li2CO3的形成反应可能为:
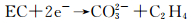
|
(16) |
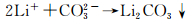
|
(17) |
ROCO2Li的形成反应为:
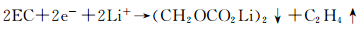
|
(18) |
ROLi的形成反应:
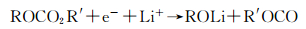
|
(19) |
其中R和R′可以是甲基或乙基。PEO可能为烷氧基和溶剂分子聚合的产物。LiF的形成可能是PF6-的直接还原:
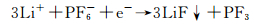
|
(20) |
或PF6-和杂质H2O的还原:
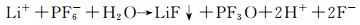
|
(21) |
首次循环后Si电极表面的SEI膜中出现了LiF,而C-Si电极上没有检测到LiF。这可能是由于Si和C-Si颗粒表面的化学性质不同,Si颗粒表面的氧化硅层含有—OH官能团,易吸湿、吸附水分,C包覆在950℃下完成,导致颗粒表面转化为疏水层,不易将水分引入电池中,因此LiF的含量降低。
两个电极的主要不同点是Si电极表面的SEI膜含有氟化硅和C组分,SiO2(而不是Si)易被HF侵蚀:
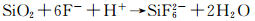
|
(22) |
反应(22)产生H2O,然后与PF6-反应生成更多的HF,合并反应(21)和(22),总反应为:
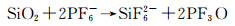
|
(23) |
SiF62-可能在电解液中溶解、扩散,也可能与Li+进一步反应生成Li2SiF6。由于C-Si电极表面没有原生氧化层,所以没有生产Si的氟化组分。
3.3 钝化膜对硅负极电化学性能的影响Si在嵌脱锂过程中伴随着巨大的体积变化,导致活性物质破碎,失去电接触,加剧了容量的衰减。活性材料表面反复破碎带来另一个非常重要的问题就是SEI膜不稳定。Radvanyi等[52]和Michan等[53]通过对纳米硅电极容量衰减原因的分析,发现容量损失与SEI的增长密切相关。在循环过程中,颗粒表面SEI膜不断生成、颗粒聚集以及SEI膜从颗粒表面脱落填充电极孔隙的综合作用导致电极密度增加。电极曲折度的增加阻碍了锂离子向体相的扩散,锂离子扩散主要集中在表面,嵌锂深度降低。SEI膜的持续生成导致不可逆容量的不断消耗,此外由于SEI膜脱落堵孔造成的电极曲折度增加,进一步造成可逆容量下降。为了解决SEI膜不稳定的问题,近十来年,研究者们在不断开发与硅负极适配的各种电解液体系。目前,在碳酸酯类电解液中加添加剂仍然是研究的主流[54-57],同时也出现了一些新的体系,如离子液体[58],聚合物电解质[59]和高浓度锂盐电解液[60]等。
4 原位表征技术在硅基负极界面反应中的应用原位表征技术越来越多地应用到硅基负极界面反应的研究中。在硅基负极锂化机理的研究方面,原位表征技术的应用已有很多报道,如原位透射电子显微镜(in situ-TEM)[61]、中子反射(neutron reflectometry)[62-63],这些原位表征技术主要是用于了解应力、扩散和粒子形态学对晶体和非晶硅反应的动力学和热力学相互关联的影响,有助于寻找具有良好电化学性能的最佳结构、形态和循环条件。
更重要的是,一些原位表征技术如原子力显微技术(AFM)、X射线反射率(XRR)和漫反射红外傅里叶变换光谱学(DRIFTS)开始应用于硅基负极表面SEI膜形成过程的研究,使得我们对SEI膜的形成过程有了更深入的认识,有助于通过设计改善SEI膜性质来提高硅负极的电化学性能。Tokranov等[64]采用原位AFM来检测SEI膜在硅电极表面的形成过程,测试结果表明最初SEI的快速形成能够在大量Li嵌入Si之前稳定下来,并且稳定速率与表面性质有关。此外,最初的循环条件也对形成的SEI膜的性质有重要影响,更快的反应速率导致更平滑,更薄的SEI薄膜。Yoon等[65]采用AFM在两种电解液1.2mol·L-1 LiPF6-EC和1.2mol·L-1 LiPF6-PC中研究了电解液组分对SEI膜演化的影响。据观察,SEI的形成主要发生在第一次锂化过程中,在EC和PC电解液中SEI膜的最大厚度分别约为17nm和10nm。Huang等[66]报道了一种原位电化学原子力显微镜(EC-AFM)方法直接观察硅电极表面形貌,并分析微米级(micron-Si)和纳米级(nano-Si)硅电极的杨氏模量。结果显示微米硅电极在储锂过程中巨大的体积变化,导致SEI表面破裂和持续生长厚而软的SEI膜。而纳米硅电极表面能够形成薄且稳定的SEI膜。杨氏模量值表明纳米硅电极的SEI膜比微米硅电极的SEI膜更硬。非原位XPS分析表明,纳米硅电极主要由无机成分组成,尤其是LiF和可能有助于提高硬度的碳酸盐类物质。由此可知,无机成分可能有助于建立具有良好杨氏模量的SEI膜以缓冲体积膨胀的硅。Yohannes等[67]采用原位DRIFT光谱技术对比分析了在1mol·L-1 LiPF6/EC:EMC(1:2):VC(2%)和1mol·L-1 LiPF6/EC:EMC(1:2):VC(2%)+FEC(10%)中硅电极表面SEI膜的形成和演化过程。结果表明没有添加FEC的电解液中,SEI膜主要由poly(VC),聚碳酸酯和碳酸锂组成。而添加10%FEC的电解液中,形成poly(VC/FEC)、聚碳酸酯、碳酸锂、烷氧基化合物和烷基磷酸酯。且添加FEC也导致poly(VC/FEC)含量的增加。这种成分变化,使得添加FEC阻碍了二次放电过程中溶剂和盐阴离子的还原分解。基于以上文献报道可知,原位表征技术在SEI膜研究中的应用,使我们能够获得SEI膜形成和性质等的直接信息,如SEI膜厚度、机械强度以及SEI膜破裂、再生成的演化过程等,有助于将SEI膜组成及性质与硅基负极的电化学性质关联,能够为SEI膜的改善、优化及设计提供指导意义。
5 结束语硅材料的储锂机制为合金化机制,储锂过程中伴随着硅表面原生氧化层的反应和钝化膜的生成。Li-Si合金化包括固溶反应和加成反应两种类型,反应类型受硅材料尺寸和充放电机制的影响,表现出不同的合金化过程。在硅材料嵌脱锂过程中,硅表面氧化层会发生两类反应,一是电化学还原,二是与电解液还原产物进一步发生化学反应,这个过程中存在容量的不可逆损耗,导致硅材料低的容量和低的库仑效率,但有利于提高硅材料的循环稳定性。电解液在硅表面的还原分解形成的钝化膜与石墨材料表面相似,其成分强烈依赖于电极电位。首次循环以及后续循环过程中电解液在硅表面还原形成钝化膜、表面氧化层的还原以及锂在活性物质硅中的不可逆嵌入均导致容量损失,而硅基负极在储锂过程中界面不稳定是造成硅基负极容量快速衰减的根本原因。因此,硅基负极材料表面与界面的调控包括材料的结构设计和电解液的优化是当前的研究热点。在材料结构设计方面已经取得了可观的成效,而电解液的优化还有待进一步提升,这就要求对电解液在硅基负极材料表面的反应进一步开展深入研究,从定性研究深入至定量的研究将成为下一步的研究重点及难点,其研究结果将对钝化膜的定量控制具有指导意义。
| [1] | WEN C J, HUGGINS R A. Chemical diffusion in intermediate phases in the lithium-silicon system[J]. Journal of Solid State Chemistry, 1981, 37 (3): 271–278. DOI: 10.1016/0022-4596(81)90487-4 |
| [2] | OBROVAC M N, CHRISTENSEN L. Structural changes in silicon anodes during lithium insertion/extraction[J]. Electrochemical and Solid State Letters, 2004, 7 (5): A93–A96. DOI: 10.1149/1.1652421 |
| [3] | HATCHARD T D, DAHN J R. In situ XRD and electrochemical study of the reaction of lithium with amorphous silicon[J]. Journal of the Electrochemical Society, 2004, 151 (6): A838–A842. DOI: 10.1149/1.1739217 |
| [4] | OBROVAC M N, KRAUSE L J. Reversible cycling of crystalline silicon powder[J]. Journal of the Electrochemical Society, 2007, 154 (2): A103–A108. |
| [5] | DING N, XU J, YAO Y X, et al. Determination of the diffusion coefficient of lithium ions in nano-Si[J]. Solid State Ionics, 2009, 180 (2/3): 222–225. |
| [6] | XIE J, IMANISHI N, ZHANG T, et al. Li-ion diffusion in amorphous Si films prepared by RF magnetron sputtering:a comparison of using liquid and polymer electrolytes[J]. Materials Chemistry and Physics, 2010, 120 (2/3): 421–425. |
| [7] | PHILIPPE B, DEDRYVÈRE R, GORGOI M, et al. Role of the LiPF6 salt for the long-term stability of silicon electrodes in Li-ion batteries-a photoelectron spectroscopy study[J]. Chemistry of Materials, 2013, 25 (3): 394–404. |
| [8] | PHARR M, ZHAO K, WANG X, et al. Kinetics of initial lithiation of crystalline silicon electrodes of lithium-Ion batteries[J]. Nano Letters, 2012, 12 (9): 5039–5047. DOI: 10.1021/nl302841y |
| [9] | FU K, YILDIZ O, BHANUSHALI H, et al. Aligned carbon nanotube-silicon sheets:a novel nano-architecture for flexible lithium ion battery electrodes[J]. Advanced Materials, 2013, 25 (36): 5109–5114. DOI: 10.1002/adma.201301920 |
| [10] | FU K, LU Y, DIRICAN M, et al. Chamber-confined silicon-carbon nanofiber composites for prolonged cycling life of Li-ion batteries[J]. Nanoscale, 2014, 6 (13): 7489–7495. DOI: 10.1039/C4NR00518J |
| [11] | CAO C, STEINRVCK H G, SHYAM B, et al. In-situ study of silicon electrode lithiation with X-ray reflectivity[J]. Nano Letters, 2016, 16 (12): 7394–7401. DOI: 10.1021/acs.nanolett.6b02926 |
| [12] | PHILIPPE B, DEDRYVERE R, ALLOUCHE J, et al. Nanosilicon electrodes for lithium-ion batteries:interfacial mechanisms studied by hard and soft X-ray photoelectron spectroscopy[J]. Chemistry of Materials, 2012, 24 (6): 1107–1115. DOI: 10.1021/cm2034195 |
| [13] | ZHANG W J. Lithium insertion/extraction mechanism in alloy anodes for lithium-ion batteries[J]. Journal of Power Sources, 2011, 196 (3): 877–885. |
| [14] | ANANI A, HUGGINS R A. Multinary alloy electrodes for solid state batteries I A phase diagram approach for the selection and storage properties determination of candidate electrode materials[J]. Journal of Power Sources, 1992, 38 (3): 351–362. DOI: 10.1016/0378-7753(92)80125-U |
| [15] | AMEZAWA K, YAMAMOTO N, TOMII Y, et al. Single-electrode Peltier heats of Li-Si alloy electrodes in LiCl-KCl eutectic melt[J]. Journal of the Electrochemical Society, 1998, 145 (6): 1986–1993. DOI: 10.1149/1.1838587 |
| [16] | BOUKAMP B A, LESH G C, HUGGINS R A. All-solid lithium electrodes with mixed-conductor matrix[J]. Journal of the Electrochemical Society, 1981, 128 (4): 725–729. |
| [17] | WU H, CUI Y. Designing nanostructured Si anodes for high energy lithium ion batteries[J]. Nano Today, 2012, 7 (5): 414–429. DOI: 10.1016/j.nantod.2012.08.004 |
| [18] | OGATA K, SALAGER E, KERR C J, et al. Revealing lithium-silicide phase transformations in nano-structured silicon-based lithium ion batteries via in situ NMR spectroscopy[J]. Nature Communications, 2014, 5 (5): 3217–3228. |
| [19] | PARK C M, KIM J H, KIM H, et al. Li-alloy based anode materials for Li secondary batteries[J]. Chemical Society Reviews, 2010, 39 (8): 3115–3141. DOI: 10.1039/b919877f |
| [20] | SHIMIZU M, USUI H, SUZUMURA T, et al. Analysis of the deterioration mechanism of Si electrode as a Li-ion battery anode using raman microspectroscopy[J]. Journal of Physical Chemistry C, 2015, 119 (6): 2975–2982. DOI: 10.1021/jp5121965 |
| [21] | LIMTHONGKUL P, JANG Y I, DUDNEY N J, et al. Electrochemically-driven solid-state amorphization in lithium-metal anodes[J]. Journal of Power Sources, 2003, 119 : 604–609. |
| [22] | LI H, HUANG X J, CHEN L Q, et al. A high capacity nano-Si composite anode material for lithium rechargeable batteries[J]. Electrochemical and Solid State Letters, 1999, 2 (11): 547–549. DOI: 10.1149/1.1390899 |
| [23] | LI J, DAHN J R. An in situ X-ray diffraction study of the reaction of Li with crystalline Si[J]. Journal of the Electrochemical Society, 2007, 154 (3): A156–A161. DOI: 10.1149/1.2409862 |
| [24] | GAO H, XIAO L, PLUMEL I, et al. Parasitic reactions in nanosized silicon anodes for lithium-ion batteries[J]. Nano Letters, 2017, 17 (3): 1512–1519. DOI: 10.1021/acs.nanolett.6b04551 |
| [25] | GHASSEMI H, AU M, CHEN N, et al. In situ electrochemical lithiation/delithiation observation of individual amorphous Si nanorods[J]. ACS Nano, 2011, 5 (10): 7805–7811. DOI: 10.1021/nn2029814 |
| [26] | LIU X H, ZHANG L Q, ZHONG L, et al. Ultrafast electrochemical lithiation of individual Si nanowire anodes[J]. Nano Letters, 2011, 11 (6): 2251–2258. DOI: 10.1021/nl200412p |
| [27] | LIU X H, ZHONG L, HUANG S, et al. Size-dependent fracture of silicon nanoparticles during lithiation[J]. ACS Nano, 2012, 6 (2): 1522–1531. DOI: 10.1021/nn204476h |
| [28] | WELL M T, RYU I, LEE S W, et al. Studying the kinetics of crystalline silicon nanoparticle lithiation with in situ transmission electron microscopy[J]. Advanced Materials, 2012, 24 (45): 6034–6041. DOI: 10.1002/adma.v24.45 |
| [29] | LIU X H, WANG J W, LIU Y, et al. In situ transmission electron microscopy of electrochemical lithiation, delithiation and deformation of individual graphene nanoribbons[J]. Carbon, 2012, 50 (10): 3836–3844. DOI: 10.1016/j.carbon.2012.04.025 |
| [30] | MISRA S, LIU N, NELSON J, et al. In situ X-ray diffraction studies of (De)lithiation mechanism in silicon nanowire anodes[J]. ACS Nano, 2012, 6 (6): 5465–5473. DOI: 10.1021/nn301339g |
| [31] | BARIS K, RANGEET B, MATHIEU M, et al. Real-time NMR investigations of structural changes in silicon electrodes for lithium-ion batteries[J]. Journal of the American Chemical Society, 2009, 131 (26): 9239–9249. DOI: 10.1021/ja8086278 |
| [32] | HUGGINS R A. Materials science principles related to alloys of potential use in rechargeable lithium cells[J]. Journal of Power Sources, 1989, 26 (1): 109–120. |
| [33] | LUO Z, FAN D, LIU X, et al. High performance silicon carbon composite anode materials for lithium ion batteries[J]. Journal of Power Sources, 2009, 189 (1): 16–21. |
| [34] | RADVANYI E, DE VITO E, PORCHER W, et al. An XPS/AES comparative study of the surface behaviour of nano-silicon anodes for Li-ion batteries[J]. Journal of Analytical Atomic Spectrometry, 2014, 29 (6): 1120–1131. DOI: 10.1039/C3JA50362C |
| [35] | XUN S, SONG X, WANG L, et al. The effects of native oxide surface layer on the electrochemical performance of Si nanoparticle-based electrodes[J]. Journal of the Electrochemical Society, 2011, 158 (12): A1260–A1266. DOI: 10.1149/2.007112jes |
| [36] | MCDOWELL M T, LEE S W, RYU I, et al. Novel size and surface oxide effects in silicon nanowires as lithium battery anodes[J]. Nano Letters, 2011, 11 (9): 4018–4025. DOI: 10.1021/nl202630n |
| [37] | GUO B, SHU J, WANG Z, et al. Electrochemical reduction of nano-SiO2 in hard carbon as anode material for lithium ion batteries[J]. Electrochemistry Communications, 2008, 10 (12): 1876–1878. DOI: 10.1016/j.elecom.2008.09.032 |
| [38] | KIM T, PARK S, OH S M. Solid-state NMR and electrochemical dilatometry study on Li+ uptake/extraction mechanism in SiO electrode[J]. Journal of the Electrochemical Society, 2007, 154 (12): A1112–A1117. DOI: 10.1149/1.2790282 |
| [39] | GRAETZ J, AHN C C, YAZAMI R, et al. Highly reversible lithium storage in nanostructured silicon[J]. Electrochemical and Solid State Letters, 2003, 6 (9): A194–A197. DOI: 10.1149/1.1596917 |
| [40] | SAINT J, MORCRETTE M, LARCHER D, et al. Towards a fundamental understanding of the improved electrochemical performance of silicon-carbon composites[J]. Advanced Functional Materials, 2007, 17 (11): 1765–1774. DOI: 10.1002/(ISSN)1616-3028 |
| [41] | LEE Y M, LEE J Y, SHIM H T, et al. SEI layer formation on amorphous si thin electrode during precycling[J]. Journal of the Electrochemical Society, 2007, 154 (6): A515–A519. DOI: 10.1149/1.2719644 |
| [42] | SUN Q, ZHANG B, FU Z W. Lithium electrochemistry of SiO2 thin film electrode for lithium-ion batteries[J]. Applied Surface Science, 2008, 254 (13): 3774–3779. DOI: 10.1016/j.apsusc.2007.11.058 |
| [43] | SCHRODER K W, DYLLA A G, HARRIS S J, et al. Role of surface oxides in the formation of solid-electrolyte interphases at silicon electrodes for lithium-ion batteries[J]. ACS Applied Materials & Interfaces, 2014, 6 (23): 21510–21524. |
| [44] | MITRA A, RIMSTIDT J D. Solubility and dissolution rate of silica in acid fluoride solutions[J]. Geochimica et Cosmochimica Acta, 2009, 73 (23): 7045–7059. DOI: 10.1016/j.gca.2009.08.027 |
| [45] | HUBAUD A A, YANG Z Z, SCHROEDER D J, et al. Interfacial study of the role of SiO2 on Si anodes using electrochemical quartz crystal microbalance[J]. Journal of Power Sources, 2015, 282 : 639–644. DOI: 10.1016/j.jpowsour.2015.02.006 |
| [46] | DIRICAN M, LU Y, FU K, et al. SiO2-confined silicon/carbon nanofiber composites as an anode for lithium-ion batteries[J]. RSC Advances, 2015, 5 (44): 34744–34751. DOI: 10.1039/C5RA03129J |
| [47] | CHAN C K, RUFFO R, HONG S S, et al. Surface chemistry and morphology of the solid electrolyte interphase on silicon nanowire lithium-ion battery anodes[J]. Journal of Power Sources, 2009, 189 (2): 1132–1140. |
| [48] | XUN S, SONG X, GRASS M E, et al. Improved initial performance of Si nanoparticles by surface oxide reduction for lithium-ion battery application[J]. Electrochemical and Solid State Letters, 2011, 14 (5): A61–A63. DOI: 10.1149/1.3559765 |
| [49] | WU H, CHAN G, CHOI J W, et al. Stable cycling of double-walled silicon nanotube battery anodes through solid-electrolyte interphase control[J]. Nature Nanotechnology, 2012, 7 (5): 309–314. |
| [50] | GOODENOUGH J B, KIM Y. Challenges for rechargeable Li batteries[J]. Chemistry of Materials, 2010, 22 (3): 587–603. DOI: 10.1021/cm901452z |
| [51] | YEN Y C, CHAO S C, WU H C, et al. Study on solid-electrolyte-interphase of Si and C-coated Si electrodes in lithium cells[J]. Journal of the Electrochemical Society, 2009, 156 (2): A95–A102. |
| [52] | RADVANYI E, PORCHER W, DE VITO E, et al. Failure mechanisms of nano-silicon anodes upon cycling:an electrode porosity evolution model[J]. Physical Chemistry Chemical Physics, 2014, 16 (32): 17142–17153. DOI: 10.1039/C4CP02324B |
| [53] | MICHAN A L, DIVITINI G, PELL A J, et al. Solid electrolyte interphase growth and capacity loss in silicon electrodes[J]. Journal of the American Chemical Society, 2016, 138 (25): 7918–7931. DOI: 10.1021/jacs.6b02882 |
| [54] | HOROWITZ Y, STEINRUVCK H G, HAN H L, et al. Fluoroethylene carbonate induces ordered electrolyte interface on silicon and sapphire surfaces as revealed by sum frequency generation vibrational spectroscopy and X-ray reflectivity[J]. Nano Letters, 2018, 18 (3): 2105–2111. DOI: 10.1021/acs.nanolett.8b00298 |
| [55] | HOROWITZ Y, HAN H L, SOTO F A, et al. Fluoroethylene carbonate as a directing agent in amorphous silicon anodes:electrolyte interface structure probed by sum frequency vibrational spectroscopy and ab initio molecular dynamics[J]. Nano Letters, 2017 . DOI: 10.1021/acs.nanolett.7b04688 |
| [56] | VEITH G M, DOUCET M, SACCI R L, et al. Determination of the solid electrolyte interphase structure grown on a silicon electrode using a fluoroethylene carbonate additive[J]. Scientific Reports, 2017, 7 (1): 6326. DOI: 10.1038/s41598-017-06555-8 |
| [57] | SCHIELE A, BREITUNG B, HATSUKADE T, et al. The critical role of fluoroethylene carbonate in the gassing of silicon anodes for lithium-ion batteries[J]. ACS Energy Letters, 2017, 2 (10): 2228–2233. DOI: 10.1021/acsenergylett.7b00619 |
| [58] | HEIST A, PIPER D M, EVANS T, et al. Self-contained fragmentation and interfacial stability in crude micron-silicon anodes[J]. Journal of the Electrochemical Society, 2018, 165 (2): A244–A250. |
| [59] | KIL E H, CHOI K H, HA H J, et al. Imprintable, bendable, and shape-conformable polymer electrolytes for versatile-shaped lithium-ion batteries[J]. Advanced Materials, 2013, 25 (10): 1395–1400. DOI: 10.1002/adma.v25.10 |
| [60] | CHANG Z H, WANG J T, WU Z H, et al. The electrochemical performance of silicon nanoparticles in concentrated electrolyte[J]. Chem Sus Chem, 2018, 11 : 1–11. DOI: 10.1002/cssc.v11.1 |
| [61] | MCDOWELL M T, RYU I, LEE S W, et al. Studying the kinetics of crystalline silicon nanoparticle lithiation with in situ transmission electron microscopy[J]. Advanced Materials, 2012, 24 (45): 6034–6041. DOI: 10.1002/adma.v24.45 |
| [62] | HVGER E, STAHN J, SCHMIDT H. Neutron reflectometry to measure in situ Li permeation through ultrathin silicon layers and interfaces[J]. Journal of the Electrochemical Society, 2015, 162 (13): A7104–A7109. DOI: 10.1149/2.0131513jes |
| [63] | JERLIU B, HVGER E, HORISBERGER M, et al. Irreversible lithium storage during lithiation of amorphous silicon thinfilm electrodes studied by in-situ neutron reflectometry[J]. Journal of Power Sources, 2017, 359 : 415–421. DOI: 10.1016/j.jpowsour.2017.05.095 |
| [64] | TOKRANOV A, SHELDON B W, LI C, et al. In situ atomic force microscopy study of initial solid electrolyte interphase formation on silicon electrodes for Li-ion batteries[J]. ACS Applied Materials & Interfaces, 2014, 6 (9): 6672. |
| [65] | YOON I, ABRAHAM D P, LUCHT B L, et al. In situ measurement of solid electrolyte interphase evolution on silicon anodes using atomic force microscopy[J]. Advanced Energy Materials, 2016, 6 (12): 1600099. DOI: 10.1002/aenm.201600099 |
| [66] | HUANG S, CHEONG L Z, WANG S, et al. In situ study of surface structure evolution of silicon anodes by electrochemical atomic force microscopy[J]. Applied Surface Science, 2018, 452 : 67–74. DOI: 10.1016/j.apsusc.2018.05.020 |
| [67] | YOHANNES Y B, WU N L, LIN S D. In situ analysis of solid electrolyte interface over Si based anodes using diffuse reflectance infrared fourier transform spectroscopy[J]. ECS Transactions, 2017, 77 (1): 71–76. |