 2022, Vol. 43
2022, Vol. 43文章信息
- 陶必林, 王钰婷, 李忠奇, 吴继周, 王建明.
- Tao Bilin, Wang Yuting, Li Zhongqi, Wu Jizhou, Wang Jianming
- 基于全基因组测序估算结核分枝杆菌分子钟
- Estimation of molecular clock of Mycobacterium tuberculosis based on whole genome sequencing data
- 中华流行病学杂志, 2022, 43(9): 1462-1468
- Chinese Journal of Epidemiology, 2022, 43(9): 1462-1468
- http://dx.doi.org/10.3760/cma.j.cn112338-20220408-00272
-
文章历史
收稿日期: 2022-04-08




2. 南京医科大学姑苏学院流行病学系, 南京 211166
2. Department of Epidemiology, Gusu School, Nanjing Medical University, Nanjing 211166, China
利用基因组测序数据推断种群进化史是进化生物学研究的重要方向[1]。自1998年完成结核分枝杆菌(Mycobacterium tuberculosis,MTB)参考序列H37Rv测序以来[2],全基因组测序(whole genome sequencing,WGS)已被广泛应用于MTB遗传多样性及进化研究[3]。基于分子钟(molecular clock)的系统发育分析可以估计MTB分支何时引入某一地区和谱系分化时间,推测最近共同祖先(most recent common ancestor,MRCA)[4-9],确定耐药分支起源和扩散时间[10-11],探讨菌株传播与人口迁徙、活动的关联[6, 9, 12]等。虽然古DNA(ancient DNA,aDNA)已被应用于研究细菌进化史[13-14],但迄今尚缺乏MTB的aDNA全基因组数据。因此,主要通过贝叶斯进化分析抽样树(Bayesian evolutionary analysis sampling trees,BEAST)构建具有MRCA校准时间的历史基因组系统发育树来推算MTB分子钟。目前,BEAST主要用于研究MTB的长期历史进化史[4-12]。但是,了解短期临床治疗过程中MTB的微进化对于理解结核病的病程发展也十分重要。微生物复制繁殖过程中,基因组通常以恒定的速率发生突变。例如,传代时间较短的细菌往往具有较高的分子进化速率[15]。因此,单位时间内基因组发生突变的数量可用于计算“分子钟”来估算细菌传代时间(或繁殖速率)。当MTB处于生长繁殖状态时,MTB具有较快的分子钟速率;反之,MTB处于潜伏或休眠等低/无生长繁殖状态时,则具有较慢的分子钟速率[16]。目前已有研究利用MTB体外培养实验和猕猴潜伏感染模型[17]、家庭传播潜伏感染[16]估算MTB分子钟。本研究利用基于全基因组测序数据,通过感染者内源性激活期间基因组位点突变率分析,开展MTB分子钟研究。
资料与方法1. 数据收集:采用[(tuberculosis)AND(relapse)] AND(whole genome sequence)组合词,检索PubMed数据库并下载全文,筛选2022年1月1日前有关MTB内源性激活复发研究,获取其上传WGS数据到欧洲核酸档案(https://www.ebi.ac.uk/ena/browser/home)或NCBI序列读取档案(https://trace.ncbi.nlm.nih.gov/Traces/sra/)平台上的BioProject ID,利用SRA Toolkit 2.11.2软件(https://github.com/ncbi/sra-tools)批量下载WGS数据库。登录NCBI SRA Run Selector网站(https://www.ncbi.nlm.nih.gov/Traces/study/)下载Metadata数据,提取每对初治-复发MTB样本对应的单核苷酸多态性(single nucleotide polymorphism,SNP)差异性以及菌株分离日期等信息。
2. MTB间遗传距离计算:对于无法获取配对菌株间SNP差异的样本,通过以下步骤计算获得:①低质量序列过滤和接头序列去除。原始测序数据采用fastp-0.20.1软件(https://github.com/OpenGene/fastp)进行质控,得到有效数据。②参考序列比对。采用BWA-0.7.17软件(https://github.com/lh3/bwa)将质控后的有效数据比对到MTB标准参考基因组H37Rv(NC_000962.3)[2],进行测序深度和覆盖率统计。③遗传距离矩阵计算。通过与H37Rv比对,生成Bam格式的结果文件,采用Freebayes-1.3.6软件(https://github.com/freebayes/freebayes)鉴定突变位点,进行变异检测和注释,采用SnpEff-5.1软件(https://github.com/pcingola/SnpEff)获得目标基因组相对于H37Rv的SNP、InDel等一系列变异信息,采用snippy-4.6.0软件(https://github.com/tseemann/snippy)生成Aln格式的多序列比对文件,运用snp-dists-0.7.0软件(https://github.com/tseemann/snp-dists)将Aln格式文件转换成MTB间SNP距离矩阵。
3. 复发类型判断:结核病复发可能是由内源性激活(endogenous relapse)导致[18],也可能由外源性再次感染(exogenous reinfection)引起[19]。本研究将初治-复发样本全基因组遗传差异≤12个SNP判断为内源性激活复发,否则判定为外源性再次感染[20]。
4. MTB分子钟估算:以月为单位衡量初治-复发的时间间隔t,假设不同繁殖速率/传代时间时,MTB平均突变率[17]:
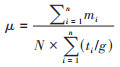
式中μ:突变率;mi:所有n对初治-复发配对MTB中,第i对MTB间SNP差异;N:MTB有效基因组大小[样本平均测序覆盖率C=99.3%,参考基因组大小L=4 411 532个碱基对(bp),N=C×L];g:传代时间,设定为18~318 h;ti:第i对初治-复发配对MTB时间间隔(月),t以月为单位,故ti/g=(365/12)×24×t/g=30.4×24×t/g。由于MTB基因组突变率低且随机发生,服从Poisson分布[16-17],因此运用Poisson回归模型拟合初治-复发时间间隔与突变率的关系。
5. 统计学分析:使用R 4.1.2软件中基础包{stats}的scatter.smooth()函数绘制初治-复发时间间隔与MTB累积SNP差异数量的散点图及loess平滑拟合曲线。使用基础包{stats}的glm()函数设定概率分布为Poisson分布,构建Poisson回归模型。检验水平α=0.05。
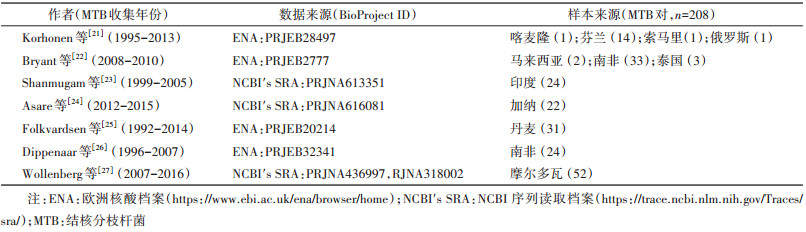
结果1. 初治-复发配对MTB样本:检索获得含初治-复发时间间隔与原始WGS数据的文献7篇,根据BioProject ID,追踪其原始WGS数据库。综合文献报告和Metadata数据,提取初治-复发配对MTB样本时间间隔(月)。依据两次样本间遗传距离判断是否为内源性激活复发,最终共纳入208对初治-复发配对样本进行分子钟分析。样本分别来源于南非(57)、摩尔多瓦(52)、丹麦(31)、印度(24)、加纳(22)、芬兰(14)、泰国(3)、马来西亚(2)、喀麦隆(1)、索马里(1)和俄罗斯(1)(表 1)。
2. 初治-复发时间间隔与配对样本间SNP差异:根据时间间隔分组进行分析[16](表 2)。依据分组方式1:超过60%在2年内发生内源性激活(129株,62.02%),初治-复发时间间隔的M(Q1,Q3)为11.73(8.40,17.47)个月,差异SNP的M(Q1,Q3)为0(0,2);在2~14年内发生内源性激活的MTB占37.98%,初治-复发时间间隔M(Q1,Q3)为44.37(34.80,68.27)个月,差异SNP的M(Q1,Q3)为2(1,5)。依据分组方式2:随着内源性激活时间间隔延长,配对MTB样本比例也逐渐减少。1年内激活复发比例最高(74株,35.58%),1~2年内激活复发菌株数为55株(26.44%)。2~、3~5年激活复发比例相同(25株,12.02%),7~14年激活复发比例最低,仅4.81%。差异SNP数在前5年呈上升趋势,0~、1~、2~和3~年差异SNP的M(Q1,Q3)分别为0(0,1)、1(0,3)、2(1,3)和3(1,5);复发时间间隔 > 5年时,差异SNP数呈下降趋势,5~、7~14年差异SNP的M(Q1,Q3)分别为3(1,6)和2(1,4)。见表 2。

采用loess平滑曲线拟合初治-复发配对样本时间间隔(t个月)与SNP差异的关系,时间间隔在42个月内,差异SNP随着时间间隔增加呈快速上升趋势;随着间隔时间继续延长,差异SNP数增加不明显,呈相对平稳状态(图 1)。

|
| 图 1 结核分枝杆菌内源性激活复发时间间隔与差异单核苷酸多态性(SNP)数的loess平滑拟合曲线 |
3. 内源性复发时间间隔与MTB突变率:研究发现,体外培养MTB对数生长期的传代时间约为18 h[28],假设体内MTB传代时间也为18 h。本研究中,MTB的WGS平均覆盖率为99.3%,基因组大小为4 411 532 bp,按每月30.4 d,每天24 h,计算可得复发时间间隔为t个月时,通过繁殖传代共产生bp数量为0.993×4 411 532×30.4×24×t÷18=177 562 398t。运用Poisson回归模型估计突变率,使用初治-复发间隔月份t为自变量,log(177 562 398t)为offset项。结果显示,t的回归系数β=-0.016(95%CI:-0.019~-0.013,P < 0.001),提示随着复发时间间隔延长,MTB突变率呈下降趋势。0~2年内激活过程中突变率(6.47×10-10,95%CI:5.59×10-10~7.44×10-10)明显高于2年后复发的突变率(3.27×10-10,95%CI:2.88×10-10~3.69×10-10)。对初治-复发时间间隔进一步细化分组,0~1年突变率为7.10×10-10(95%CI:5.66×10-10~8.76×10-10);1~、2~、3~、5~、7~14年突变率分别为6.06×10-10(95%CI:4.99×10-10~7.28×10-10)、4.24×10-10(95%CI:3.25×10-10~5.42×10-10)、5.34×10-10(95%CI:4.38×10-10~6.43×10-10)、2.59×10-10(95%CI:2.00×10-10~3.30×10-10)、1.26×10-10(95%CI:8.19×10-11~1.83×10-10),同样观察到随着时间间隔延长,MTB突变率呈下降的趋势(表 3)。

通过Poisson模型进行曲线拟合,可以发现MTB突变率随着时间间隔增加而降低,且突变率下降的趋势随着初治-复发时间间隔增加而变得平缓(图 2)。

|
| 图 2 宿主人类体内结核分枝杆菌内源性激活复发时间间隔与结核分枝杆菌突变率的关系 |
4. MTB突变率与传代时间的关系:分析不同传代时间(18~318 h)MTB突变率变化趋势[17]。结果发现,2~14年内突变率明显降低,约为0~2年内突变率的一半(表 3,图 3)。随着MTB传代时间延长,MTB突变率均呈上升趋势(图 3)。

|
| 图 3 不同传代时间下内源性激活期间结核分枝杆菌突变率 |
基于分子钟的系统发育分析已被广泛运用于推断MTB基因组进化史[4-9],通过BEAST构建MRCA校准时间的基因组系统发育树可以推算MTB长期的历史进化过程[4-12]。而基于短期的临床案例中MTB突变率研究则可以了解MTB微进化或特殊临床期菌株繁殖情况。宿主感染MTB后,可能的结局包括:①MTB被先天和/或获得性免疫清除;②潜伏感染(最常见);③快速进展发病;④缓慢进展发病,通过早期和亚临床阶段发展为活动性结核病;⑤处于早期和亚临床状态的循环期[29]。MTB生长繁殖特点与致病性密切相关,宿主体内MTB生长繁殖较快时毒力及致病性较高,更易出现活动性结核症状[29-30]。因此结核病病程或疾病严重程度可以借助MTB分子钟量化识别MTB繁殖特点进行推断,对实现指导结核精准防治具有重要的临床和公共卫生学意义。
本研究发现,初治-复发间隔时间在2年内,MTB突变率(分子钟速率)相对较高,可称为“早期内源性激活”。随着间隔时间延长,MTB逐渐转变为低突变率(低分子钟速率)状态,可称之为“晚期内源性激活”。本研究从病原体微观角度揭示了临床实践中观察到的宏观现象,即大多数复发出现于初治后2年内,尤其第1年复发风险最高[31]。这可用本研究观察到的“早期内源性激活”解释。2年后复发风险下降的临床现象与“晚期内源性激活”机制吻合。
实验室条件下定义的突变率与自然界中随时间累积的突变率相结合,已被用于估计各种细菌的复制率[32]。根据人工培养基中MTB对数期生长时间[17],假设内源性激活传代时间为18 h,本研究发现MTB突变率随复发间隔时间延长而降低。由于无法直接测量机体内MTB确切传代时间,本研究进一步考虑了各种可能传代时间(18~318 h)下的MTB突变率。结果显示,无论是“早期内源性激活”还是“晚期内源性激活”阶段,随着传代时间延长,MTB突变率均呈上升趋势。造成这一现象的原因是内源性激活期间经历的传代次数与传代时间成反比,故推算的MTB突变率增加使得在固定的复发时间间隔内仍然观测到相同数量SNP。
Colangeli等[16]基于24对样本的研究发现,MTB突变率随着潜伏期延长而显著降低(β=-0.053,95%CI:-0.078~-0.029)。若传代时间为18 h,潜伏期在0~2年内的MTB突变率为8.36×10-10(95%CI:3.71×10-10~1.88×10-9),略高于本研究;潜伏期在2~6年内的MTB突变率为3.81×10-10(95%CI:1.70×10-10~8.53×10-9)。但Colangeli等[16]未发现指示病例与其匹配的续发病例菌株间SNP差异随着潜伏期延长而增加,可能与该研究中MTB潜伏感染期间未接受抗结核药物影响所致。
林楠[33]基于BEAST构建基因组系统发育树,以MTB非洲分支L5与L6谱系的MRCA先验分布[4]为校准时间,选用HKY+Γ为碱基替换模型,在对数正态松弛钟(lognormal relaxed clock)下进行推算,发现MTB每个位点每年突变率约2.59×10-9,与Comas等[4]基于BEAST预测的突变率基本一致(2.58×10-9)。然而,Ford等[17]构建猕猴潜伏感染模型发现,MTB每个位点每年突变率为1.17×10-7(95%CI:2.39×10-8~3.40×10-7),在研究L2与L4谱系体外实验菌株突变率差异时,发现每个位点每年突变率为6.80×10-8~1.13×10-7。经单位换算,本研究中,初治-复发时间间隔0~2年时MTB每个位点每年突变率为3.15×10-7(95%CI:2.72×10-7~3.62×10-7);2~14年时突变率为1.59×10-7(95%CI:1.40×10-7~1.80×10-7),略高于Ford等[17]的猕猴潜伏感染模型和体外实验结果。
研究发现,基于长期的基因组系统发育分析预测的突变率明显低于短期的体外实验、猕猴和人类潜伏感染模型及本研究中人类内源性激活案例,导致这一现象的原因可能有:①MTB在长期进化中受到负向选择(purifying selection)压力[34],有害突变在长期进化过程中逐渐消失,存在大片段缺失[35],导致长期、跨度达数万年计算出的突变率低于短期突变率;②人类不断迁徙,奠基者效应引发遗传漂变(genetic drift)[4, 9, 36],使得一些低频突变未被保留而降低突变率;③人型MTB与人类免疫系统有着高度适应性,MTB在刚感染宿主的情况下,受到较强正向选择(positive selection)而提高突变率[37]。然而,第三点因素的影响应该不足以导致MTB突变率数十倍差异,因此第一种因素很可能是造成突变率差异较大的主要原因。综上所述,根据MTB实验菌株所计算的突变率应该更接近于临床MTB的实际突变率,适合于推断MTB的短期微进化过程;而林楠[33]基于系统发育树分析计算的突变率则适合于推断MTB的长期历史进化过程。
当MTB处于较高的复制繁殖状态时,固定时间内MTB具有较多的繁殖传代次数,这一活跃生理状态表明此时期MTB具有较高的分子钟速率,意味着宿主体内累积更多的MTB,使患者快速发展为活动性结核而出现典型临床症状、影像学特征及细菌学检测阳性[16, 29]。因此利用MTB分子钟可以预测特定时期内的细菌生长繁殖情况,继而对宿主的发病风险进行预测,寻找内源性激活风险窗口期,为开展针对性预防提供理论支撑。
对一线抗结核药敏感的结核病患者在6个月的治疗后,仍有约5%的患者复发[38]。复发率高低意味着抗结核治疗预后的好坏,同时复发患者也可能成为新的传染源[39-40]。结核病例体内MTB的复制繁殖受阻碍细胞生长途径的药物(异烟肼/利福平)影响,但处于非复制繁殖或低复制繁殖状态的MTB对这些抗结核药耐受性较强[41-42]。识别患者MTB的繁殖活动状态,就能有针对性地预防性治疗,有效防止或降低复发风险[43]。
本研究利用内源性激活复发期间MTB基因组位点突变来估计MTB分子钟,揭示了内源性激活复发期间MTB繁殖活跃程度变化情况。前2年内MTB分子钟速率最高,随着时间推移,分子钟速率逐渐降低,意味着MTB的复制繁殖活性随着时间的推移逐渐降低,即体内未被完全清除的MTB数量处于较低水平。因此,本研究结果显示,预防MTB内源性激活复发的时间应越早越好。
利益冲突 所有作者声明无利益冲突
作者贡献声明 陶必林:查阅文献、测序数据下载、数据整理、统计分析绘图、文章撰写;王钰婷:数据整理、论文修改;李忠奇、吴继周:数据整理;王建明:研究设计、获取研究经费、论文指导、论文修改、论文审核
| [1] |
Kimura M. Evolutionary rate at the molecular level[J]. Nature, 1968, 217(5129): 624-626. DOI:10.1038/217624a0 |
| [2] |
Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence[J]. Nature, 1998, 393(6685): 537-544. DOI:10.1038/31159 |
| [3] |
Riviere E, Heupink TH, Ismail N, et al. Capacity building for whole genome sequencing of Mycobacterium tuberculosis and bioinformatics in high TB burden countries[J]. Brief Bioinform, 2021, 22(4). DOI:10.1093/bib/bbaa246 |
| [4] |
Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans[J]. Nat Genet, 2013, 45(10): 1176-1182. DOI:10.1038/ng.2744 |
| [5] |
Bos KI, Harkins KM, Herbig A, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis[J]. Nature, 2014, 514(7523): 494-497. DOI:10.1038/nature13591 |
| [6] |
Merker M, Blin C, Mona S, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage[J]. Nat Genet, 2015, 47(3): 242-249. DOI:10.1038/ng.3195 |
| [7] |
Kay GL, Sergeant MJ, Zhou Z, et al. Eighteenth-century genomes show that mixed infections were common at time of peak tuberculosis in Europe[J]. Nat Commun, 2015, 6: 6717. DOI:10.1038/ncomms7717 |
| [8] |
Brynildsrud OB, Pepperell CS, Suffys P, et al. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation[J]. Sci Adv, 2018, 4(10): eaat5869. DOI:10.1126/sciadv.aat5869 |
| [9] |
Liu Q, Ma A, Wei L, et al. China's tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis[J]. Nat Ecol Evol, 2018, 2(12): 1982-1992. DOI:10.1038/s41559-018-0680-6 |
| [10] |
Eldholm V, Monteserin J, Rieux A, et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain[J]. Nat Commun, 2015, 6: 7119. DOI:10.1038/ncomms8119 |
| [11] |
Cohen KA, Abeel T, Manson Mcguire A, et al. Evolution of Extensively Drug-Resistant Tuberculosis over Four Decades: Whole Genome Sequencing and Dating Analysis of Mycobacterium tuberculosis Isolates from KwaZulu- Natal[J]. PLoS Med, 2015, 12(9): e1001880. DOI:10.1371/journal.pmed.1001880 |
| [12] |
Eldholm V, Pettersson JH, Brynildsrud OB, et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis[J]. Proc Natl Acad Sci USA, 2016, 113(48): 13881-13886. DOI:10.1073/pnas.1611283113 |
| [13] |
Bos KI, Schuenemann VJ, Golding GB, et al. A draft genome of Yersinia pestis from victims of the Black Death[J]. Nature, 2011, 478(7370): 506-510. DOI:10.1038/nature10549 |
| [14] |
Djelouadji Z, Raoult D, Drancourt M. Palaeogenomics of Mycobacterium tuberculosis: epidemic bursts with a degrading genome[J]. Lancet Infect Dis, 2011, 11(8): 641-650. DOI:10.1016/S1473-3099(11)70093-7 |
| [15] |
Thomas JA, Welch JJ, Lanfear R, et al. A generation time effect on the rate of molecular evolution in invertebrates[J]. Mol Biol Evol, 2010, 27(5): 1173-1180. DOI:10.1093/molbev/msq009 |
| [16] |
Colangeli R, Gupta A, Vinhas SA, et al. Mycobacterium tuberculosis progresses through two phases of latent infection in humans[J]. Nat Commun, 2020, 11(1): 4870. DOI:10.1038/s41467-020-18699-9 |
| [17] |
Ford CB, Lin PL, Chase MR, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection[J]. Nat Genet, 2011, 43(5): 482-486. DOI:10.1038/ng.811 |
| [18] |
Dippenaar A, de Vos M, Marx FM, et al. Whole genome sequencing provides additional insights into recurrent tuberculosis classified as endogenous reactivation by IS6110 DNA fingerprinting[J]. Infect Genet Evol, 2019, 75. |
| [19] |
Schiroli C, Carugati M, Zanini F, et al. Exogenous reinfection of tuberculosis in a low-burden area[J]. Infection, 2015, 43(6): 647-653. DOI:10.1007/s15010-015-0759-9 |
| [20] |
Yang C, Luo T, Shen X, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation[J]. Lancet Infect Dis, 2017, 17(3): 275-284. DOI:10.1016/S1473-3099(16)30418-2 |
| [21] |
Korhonen V, Smit PW, Haanpera M, et al. Whole genome analysis of Mycobacterium tuberculosis isolates from recurrent episodes of tuberculosis, Finland, 1995- 2013[J]. Clin Microbiol Infect, 2016, 22(6): 549-554. DOI:10.1016/j.cmi.2016.03.014 |
| [22] |
Bryant JM, Harris SR, Parkhill J, et al. Whole-genome sequencing to establish relapse or re-infection with Mycobacterium tuberculosis: a retrospective observational study[J]. Lancet Respir Med, 2013, 1(10): 786-792. DOI:10.1016/S2213-2600(13)70231-5 |
| [23] |
Shanmugam S, Bachmann NL, Martinez E, et al. Whole genome sequencing based differentiation between re-infection and relapse in Indian patients with tuberculosis recurrence, with and without HIV co-infection[J]. Int J Infect Dis, 2021, 113(Suppl 1): S43-47. DOI:10.1016/j.ijid.2021.03.020 |
| [24] |
Asare P, Osei-Wusu S, Baddoo NA, et al. Genomic epidemiological analysis identifies high relapse among individuals with recurring tuberculosis and provides evidence of recent household-related transmission of tuberculosis in Ghana[J]. Int J Infect Dis, 2021, 106: 13-22. DOI:10.1016/j.ijid.2021.02.110 |
| [25] |
Folkvardsen DB, Norman A, Rasmussen EM, et al. Recurrent tuberculosis in patients infected with the predominant Mycobacterium tuberculosis outbreak strain in Denmark. New insights gained through whole genome sequencing[J]. Infect Genet Evol, 2020, 80: 104169. DOI:10.1016/j.meegid.2020.104169 |
| [26] |
Dippenaar A, de Vos M, Marx FM, et al. Whole genome sequencing provides additional insights into recurrent tuberculosis classified as endogenous reactivation by IS6110 DNA fingerprinting[J]. Infect Genet Evol, 2019, 75: 103948. DOI:10.1016/j.meegid.2019.103948 |
| [27] |
Wollenberg K, Harris M, Gabrielian A, et al. A retrospective genomic analysis of drug-resistant strains of M. tuberculosis in a high-burden setting, with an emphasis on comparative diagnostics and reactivation and reinfection status[J]. BMC Infect Dis, 2020, 20(1): 17. DOI:10.1186/s12879-019-4739-z |
| [28] |
Trojanowski D, Ginda K, Pioro M, et al. Choreography of the Mycobacterium replication machinery during the cell cycle[J]. mBio, 2015, 6(1): e02125-14. DOI:10.1128/mBio.02125-14 |
| [29] |
Drain PK, Bajema KL, Dowdy D, et al. Incipient and Subclinical Tuberculosis: a Clinical Review of Early Stages and Progression of Infection[J]. Clin Microbiol Rev, 2018, 31(4). DOI:10.1128/CMR.00021-18 |
| [30] |
Tram TTB, Nhung HN, Vijay S, et al. Virulence of Mycobacterium tuberculosis Clinical Isolates Is Associated With Sputum Pre-treatment Bacterial Load, Lineage, Survival in Macrophages, and Cytokine Response[J]. Front Cell Infect Microbiol, 2018, 8: 417. DOI:10.3389/fcimb.2018.00417 |
| [31] |
Colangeli R, Jedrey H, Kim S, et al. Bacterial Factors That Predict Relapse after Tuberculosis Therapy[J]. N Engl J Med, 2018, 379(9): 823-833. DOI:10.1056/NEJMoa1715849 |
| [32] |
Gibson B, Wilson DJ, Feil E, et al. The distribution of bacterial doubling times in the wild[J]. Proc Biol Sci, 2018, 285(1880). DOI:10.1098/rspb.2018.0789 |
| [33] |
林楠. 中国结核分枝杆菌群体基因组的分析[D]. 福州: 福建农林大学, 2014.
|
| [34] |
Lee RS, Radomski N, Proulx JF, et al. Population genomics of Mycobacterium tuberculosis in the Inuit[J]. Proc Natl Acad Sci USA, 2015, 112(44): 13609-13614. DOI:10.1073/pnas.1507071112 |
| [35] |
Brosch R, Gordon SV, Marmiesse M, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex[J]. Proc Natl Acad Sci USA, 2002, 99(6): 3684-3689. DOI:10.1073/pnas.052548299 |
| [36] |
Pepperell CS, Casto AM, Kitchen A, et al. The role of selection in shaping diversity of natural M. tuberculosis populations[J]. PLoS Pathog, 2013, 9(8): e1003543. DOI:10.1371/journal.ppat.1003543 |
| [37] |
Vargas R, Freschi L, Marin M, et al. In-host population dynamics of Mycobacterium tuberculosis complex during active disease[J]. Elife, 2021, 10. DOI:10.7554/eLife.61805 |
| [38] |
Lambert ML, Hasker E, Van Deun A, et al. Recurrence in tuberculosis: relapse or reinfection?[J]. Lancet Infect Dis, 2003, 3(5): 282-287. DOI:10.1016/s1473-3099(03)00607-8 |
| [39] |
Merle CS, Fielding K, Sow OB, et al. A four-month gatifloxacin-containing regimen for treating tuberculosis[J]. N Engl J Med, 2014, 371(17): 1588-1598. DOI:10.1056/NEJMoa1315817 |
| [40] |
Gillespie SH, Crook AM, Mchugh TD, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis[J]. N Engl J Med, 2014, 371(17): 1577-1587. DOI:10.1056/NEJMoa1407426 |
| [41] |
Zhang Y. Persistent and dormant tubercle bacilli and latent tuberculosis[J]. Front Biosci, 2004, 9: 1136-1156. DOI:10.2741/1291 |
| [42] |
Gomez JE, Mckinney JD. M. tuberculosis persistence, latency, and drug tolerance[J]. Tuberculosis (Edinb), 2004, 84(1/2): 29-44. DOI:10.1016/j.tube.2003.08.003 |
| [43] |
Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis[J]. Nat Rev Immunol, 2017, 17(11): 691-702. DOI:10.1038/nri.2017.69 |