 2022, Vol. 43
2022, Vol. 43文章信息
- 梁文娟, 胡爱玲, 龙金照, 朱金钦, 段广才.
- Liang Wenjuan, Hu Ailing, Long Jinzhao, Zhu Jinqin, Duan Guangcai
- 基于不同方法的大肠埃希菌分型效果评价
- Evaluation of effect based on different typing methods in Escherichia coli
- 中华流行病学杂志, 2022, 43(8): 1321-1325
- Chinese Journal of Epidemiology, 2022, 43(8): 1321-1325
- http://dx.doi.org/10.3760/cma.j.cn112338-20220303-00167
-
文章历史
收稿日期: 2022-03-03




2. 郑州大学公共卫生学院流行病教研室, 郑州 450001
2. Department of Epidemiology, College of Public Health, Zhengzhou University, Zhengzhou 450001, China
细菌分型是疫情调查、监测和系统发育研究的重要工具,在流行病学溯源调查中发挥重要作用[1]。目前大肠埃希菌的分型方法主要有血清学分型、脉冲场凝胶电泳(pulsed field gel electrophoresis,PFGE)、多位点序列分型(multilocus sequence typing,MLST)[2]等,其中血清型长期用于致病大肠埃希菌分型,但对于新发未知菌株,在确定其血清型时,耗费时间、人力和物力;MLST是基于DNA序列的分子生物学分型技术,该法对大肠埃希菌7个管家基因adk、fumC、gyrB、icd、mdh、purA和recA进行扩增测序,将测序结果与MLST数据库进行比对获取菌株的分型结果,即序列型(sequence type,ST),该方法数据结果可用于大规模和长期流行病学监测。
成簇规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPRs)由一段不连续的重复序列和高度多变的间隔序列组成,细菌在进化过程中不断捕获更新外源可移动元件如噬菌体或质粒,形成更加动态多变间隔序列(spacer)。基于CRISPRs间隔序列的细菌分型,一定程度上不仅能反映细菌内在的遗传关系,还能显示出进化路径关系[3]。在大肠埃希菌中存在I-E型或I-F型CRISPR/Cas系统,CRISPR1、紧邻的cas和下游20 kb的CRISPR2共同组成I-E型CRISPR/Cas系统,CRISPR3及下游的cas和CRISPR4位点共同组成I-F型CRISPR/Cas系统。课题组前期发现CRISPRs的间隔序列的多态性可以作为大肠埃希菌分子分型的靶标[4],但是并未将I-F型CRISPR/Cas和CRISPR3-4纳入大肠埃希菌的分型体系,且未与MLST和血清型方法进行比较。本研究选取203株全基因组序列测序不同致病类型的大肠埃希菌,分析CRISPR/Cas的结构特征,建立CRISPRs大肠埃希菌的分型方法,并与MLST和血清分型方法比较,同时扩增实验室保存的349株大肠埃希菌的CRISPRs并测序分析,评价其分型效果。
材料与方法1. 菌株:选取GenBank中203株大肠埃希菌全基因组序列和349株实验室保存的大肠埃希菌。试验菌株主要来自2014-2015年新乡医学院第三附属医院和河南省商丘睢县腹泻哨点监测医院,所有实验室菌株均进行API鉴定。
2. 主要仪器和试剂:诊断血清购自中国宁波天润生物药业有限公司;API20E生化鉴定板条购自法国生物梅里埃公司;PTC-100型基因体外扩增仪,加拿大MJ Research公司;DYY-8C型稳压稳流定时电泳仪购自中国北京六一仪器厂;Gene snap图像扫描仪购自美国Syngene公司。
3. 203株大肠埃希菌全基因组序列分型:①CRISPRs的识别和间隔序列分析:具体方法参考文献[4]。②血清型:通过在线软件SeroTypeFinder(https://cge.cbs.dtu.dk/services/serotypeFinder/)获得。③MLST分型:通过在线软件MLST Finder(https://cge.cbs.dtu.dk/services/MLST/)获得ST。
采用辛普森指数评价3种分型方法的分辨率;采用调整兰德指数(adjusted Rand index)评价任意2种分型方法之间吻合程度。采用R 4.1.3软件计算相应指标,数值越大分别代表分辨率越高和吻合程度越强。
4. 349株实验室保存的大肠埃希菌分型:①CRISPRs和MLST的检测:采用煮沸法提取大肠埃希菌DNA,采用PCR扩增CRISPRs和大肠埃希菌7个管家基因adk、fumC、gyrB、icd、mdh、purA、recA,CRISPR1、3、4引物序列参考相关文献[5],CRISPR2和CRISPR3-4引物序列参考文献[6]。7个管家基因的引物和扩增程序参考MLST官网(http://www.mlst.net)。CRISPRs和MLST管家基因扩增产物委托生工生物工程(上海)股份有限公司进行双向测序、结果拼接。测得CRISPRs序列使用CRISPRsFinder分析,管家基因上传MLST官网获得ST。②血清学鉴定:使用诊断血清确定血清型,具体方法参照产品说明书。
结果1. 大肠埃希菌CRISPRs的间隔序列:203株大肠埃希菌全基因组序列中,有151株大肠埃希菌存在I-E型CRISPR/Cas,其中CRISPR1和CRISPR2中存在1 337条和1 216条间隔序列(各包含339条和346条独特的间隔序列);有19株存在I-F型CRISPR/Cas,其中CRISPR3和CRISPR4中存在155条和180条间隔序列(包含57条和66条独特的间隔序列);35株无I-E型和I-F型CRISPR/Cas,仅在I-F型CRISPR/Cas的位置留有一段序列,即CRISPR3-4,共发现40条间隔序列和5条独特的间隔序列。
2. 基于CRISPRs的大肠埃希菌分型:将I-E CRISPR定义为CT-Ⅰ、I-F型CRISPR定义为CT-Ⅱ、CRISPR3-4定义为CT-Ⅲ,即基于CRISPRs将大肠埃希菌分为3个型别,然后基于每个CRISPRs间隔序列构成和排列顺序,将其分为相应的亚型,203株大肠埃希菌被分为79个CT亚型,其中CT-Ⅰ型64个,CT-Ⅱ型为9个,CT-Ⅲ型为6个。
3. 评价CRISPRs对大肠埃希菌的分型效果:203株大肠埃希菌分成76个血清型和66个ST。采用辛普森指数评价3种分型方法的分辨率,结果显示CRISPRs的辛普森指数为0.936,略高于血清型(0.935)和MLST(0.933)。采用调整兰德指数评估任意2种分型方法之间吻合程度,结果显示CRISPRs和血清学的一致程度最高为0.908,CRISPRs和MLST的一致程度为0.761,血清学和MLST的一致程度最低为0.738。
部分菌株的CRISPRs亚型与ST和(或)血清型具有严格的一致性:比如CT-Ⅰ 3与O55:H7和ST335;CT-Ⅲ 1与O7:H45和ST62;CT-Ⅲ 5与ST131等。见表 1。
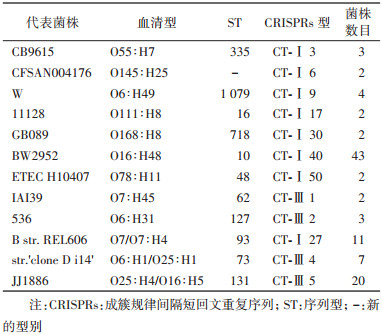
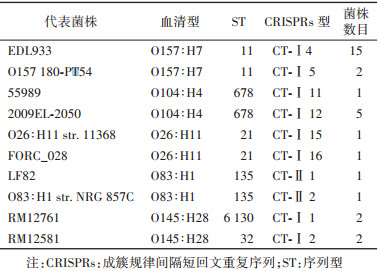
CRISPRs型能将相同血清型和(或)ST的大肠埃希菌分成不同的亚型:将大肠埃希菌O157:H7,O104:H4,O26:H11,O83:H1分成2类,分别分成CT-Ⅰ 4和CT-Ⅰ 5,CT-Ⅰ 11和CT-Ⅰ 12,CT-Ⅰ 15和CT-Ⅰ 16,CT-Ⅱ 1和CT-Ⅱ 2(表 2);将ST95菌株分为4类:CT-Ⅱ 3,CT-Ⅱ 4,CT-Ⅱ 6和CT-Ⅱ 7(表 3);将O145:H28分成2类,ST6130与CT-Ⅰ 1,ST32与CT-Ⅰ 2两类(表 2)。表现部分相同ST和(或)CRISPRs型的大肠埃希菌其血清型表现多样化(表 3)。不同血清型菌株如大肠埃希菌12009(O103:H2),11368(O26:H11)和11128(O111:H8)既具有部分相同的间隔序列,也存在不同的间隔序列,可显示进化的亲缘关系。

4. 实验验证基于CRISPRs的大肠埃希菌分型效果:收集并鉴定大肠埃希菌349株,其中134株分离于2014年河南省新乡市;215株分离于河南省睢县(2002年40株,2014年175株)。PCR扩增CRISPR1、CRISPR2、CRISPR3、CRISPR4和CRISPR3-4,其检出率分别为81.1%(283株)、94.5%(330株)、1.4%(5株)、1.4%(5株)和4.6%(16株)。PCR产物测序结果使用CRISPRsfinder或者BLAST分析间隔序列,根据CRISPR1、2的间隔序列分布预测38株O157:H7(ST11)、34株O16:H48(ST10)肠共生大肠埃希菌;根据CRISPR3-4间隔序列预测13株O25:H4(ST131)大肠埃希菌。本研究对实验室菌株进行O157:H7、O16:H48和O25:H4的血清型和MLST鉴定,结果显示存在40株O157:H7(ST11)和13株ST131菌株;根据CRISPRs的间隔序列分布预测O157:H7(ST11)和ST131的一致率分别为95.0%和100.0%,因O16:H48和O25:H4的血清凝集结果并不明显,所以未进行分析。
讨论细菌分型是识别和追溯传染源的重要流行病学工具,好的分型方法必须具备高度鉴别力和追溯性[7]。本研究结果显示,CRISPRs分辨率和与血清学分型一致程度较高,扩增实验室保存的349株大肠埃希菌的CRISPRs并测序分析,根据CRISPRs间隔序列预测O157:H7(ST11)和ST131准确率分别为95.0%和100.0%。
本研究将I-E型CRISPRs定义为CT-Ⅰ、I-F型CRISPRs定义为CT-Ⅱ、CRISPR3-4定义为CT-Ⅲ,即基于CRISPRs将大肠埃希菌分为3个型别,可涵盖目前分析的所有大肠埃希菌,基于CRISPRs可以对203株不同致病类型的大肠埃希菌分为79个CT型别。已有研究表明serotypeFinder和MLST对于识别大肠埃希菌全基因组序列血清型和ST具有很好的敏感性和特异性[8-9],分析显示203株大肠埃希菌被分为76个血清型和66个ST。
运用辛普森指数评价基于CRISPRs的大肠埃希菌分子分型效果,结果表明CRISPRs分型比血清分型及MLST方法具备更高的分辨力。Zeng等[10]在克罗诺杆菌中的研究发现与MLST方法相比,CRISPRs分型有更高的分辨率。MLST分辨率最低,通过检测大肠埃细菌的管家基因,其序列突变积累较慢,导致进化相对保守,而CRISPRs序列是细菌记录其整合外源遗传物质某个片段,其持续动态多样化的特征为分型奠定了基础。推测MLST管家基因相对比较保守,对进化的反映可能不如CRISPRs快,与Marraffini[11]认为CRISPRs序列是为细菌中进化最快的位点之一研究结果一致。血清型依据细菌表面抗原—菌体抗原(O抗原)和鞭毛抗原(H抗原),细菌进化过程中表面抗原也会经常发生变异,但其变异的速度可能没有CRISPRs快,因此分辨率不如CRISPRs高。其次,本研究使用调整兰德指数评估任意2种分型方法之间吻合程度,结果显示CRISPRs和血清学的一致程度最高,显示大肠埃希菌CRISPRs和细菌表面抗原进化的一致性。但是大肠埃希菌细菌表面抗原会导致血清学检测方法不能有效地识别变异菌株和新发未知菌株,且该方法易受到血清质量、血清交叉反应以及菌体表面其他抗原成分的影响,确定其血清型时不仅比较耗费时间、物力和人力,使可能影响检测准确度、检出率和灵敏度[12]。CRISPRs和MLST的一致程度为0.761,MLST法需要测序7条序列,相对费用较高,且因个别碱基的变化会导致型别改变,因此对测序技术要求非常严格[13]。此时用CRISPRs分型替代血清型和MLST,不仅降低工作量,还可以节省成本。更重要的是,CRISPRs型能将相同血清型和ST的大肠埃希菌分成不同的亚型:如CT-Ⅰ 4的15株O157:H7均分离于2011年之前[14-15],而CT-Ⅰ 5的2株O157:H7于2012年分离自英国;CT-Ⅰ 11的1株细菌55989来源于非洲中部艾滋病患者,属肠黏附大肠埃希菌,CT-Ⅰ 12的2株细菌分别为引起2009年和2011年的欧洲暴发菌株[16];ST21的O26:H11大肠埃希菌CT-Ⅰ 15和CT-Ⅰ 16,分别分离于2003年韩国和2001年日本[17]。CRISPRs还能将相同血清型O145:H28分成2类CT-Ⅰ 1与CT-Ⅰ 2,CT-Ⅰ 1于2007年分离于比利时,CT-Ⅰ 2于2010年分离于美国[18],由此推测基于CRISPRs的分型有可能提示会跟菌株的分离时间和地区有一定的关系,有可能在追踪传染源会更具价值。CRISPRs能将相同的ST(血清型不同)的大肠埃希菌分成亚型:如ST10的VR50全基因组序列测序显示类似肠共生大肠埃希菌,但是获得了能黏附人类膀胱的相关毒力基因[19];分离自同一地区ST95菌株SF-173、SF-166、SF-088和SF-468,分离于2007-2010年,有不同的fimH等位基因,其研究者认为跟耐药和宿主自身的适应性有关[20]。CRISPRs的间隔序列能反映出菌株间的亲缘关系。前期已发现O55:H7和O157:H7、O104:H4与肠黏附大肠埃希菌55989的关系。本研究发现产志贺毒素大肠埃希菌O26:H11、103:H2、O111:H8既有相同的间隔序列,也有不同的间隔序列,由此推测O26:H11、O103:H2、O111:H8的相同的间隔序列可能反映出经历过相同的进化环境,但在某个节点又进入不同的进化分支上。Ogura等[17]研究3株全基因测序的O26:H11、O103:H2、O111:H8大肠埃希菌,显示存在噬菌体、整合元件和携带相同或相似毒力基因的质粒,但是却有不同的进化历史,驱使各自的进化。Yang等[21]研究发现相同型别的CRISPRs位点可以出现在相同的MLST金葡菌菌株中,推测相同MLST型别的金葡菌菌株可能在遗传关系上彼此紧密相连,CRISPRs位点所在基因元件在入侵细菌基因组时,菌株对于外源入侵CRISPRs位点的接收具有一定的选择性。在大肠埃希菌中,目前并未发现可移动基因元件上存在CRISPRs,有待于进一步研究。
本研究进一步验证CRISPRs分型方法的临床应用效果,设计引物扩增并测序分析实验室349株大肠埃希菌CRISPRs的间隔序列,根据CRISPRs的间隔序列分布预测O157:H7(ST11)的准确率是95.0%,ST131的准确性达100.0%。有2株O157:H7的CRISPR1未扩增出来,其余4株O157:H7的CRISPR2的间隔序列分别4条,这种多样性的存在也许是因为菌株在进化路径不一致产生的,也提示当时分离地区存在多种亚型的大肠埃希菌O157:H7,推测其可以作为亚型分型,有待后续进一步研究。研究显示血清型O16:H48和O25:H4的血清凝集结果并不明显,提示对于K12肠共生大肠埃希菌和肠外致病菌O25:H4,其血清型并不适用。
研究显示,与血清学和MLST方法相比,基于CRISPRs的大肠埃希菌的分子分型方法呈现较好的分型效果和临床应用效果,预期可以成为大肠埃希菌分型的重要分子标志物。
利益冲突 所有作者声明无利益冲突
作者贡献声明 梁文娟:实验操作、论文撰写和经费支持;胡爱玲、龙金照、朱金钦:数据整理、统计学分析;段广才:研究指导、经费支持
| [1] |
Sabat AJ, Budimir A, Nashev D, et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance[J]. Euro Surveill, 2013, 18(4): 20380. |
| [2] |
黄涛, 山珊, 黄艳梅, 等. 大肠埃希氏菌的分型方法及其研究进展[J]. 微生物学通报, 2020, 47(3): 892-902. Huang T, Shan S, Huang YM, et al. Advances in typing methods for Escherichia coli[J]. Microbiol China, 2020, 47(3): 892-902. DOI:10.13344/j.microbiol.china.190630 |
| [3] |
Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants[J]. Nat Rev Microbiol, 2020, 18(2): 67-83. DOI:10.1038/s41579-019-0299-x |
| [4] |
梁文娟, 张荣光, 段广才, 等. 基于CRISPR/Cas的大肠埃希菌分子标志物的监测研究[J]. 中华流行病学杂志, 2016, 37(8): 1080-1086. Liang WJ, Zhang RG, Duan GC, et al. A surveillance study on CRISPR/Cas molecular biomarker in Escherichia coli[J]. Chin J Epidemiol, 2016, 37(8): 1080-1086. DOI:10.3760/cma.j.issn.0254-6450.2016.08.005 |
| [5] |
Touchon M, Charpentier S, Clermont O, et al. CRISPR distribution within the Escherichia coli species is not suggestive of immunity-associated diversifying selection[J]. J Bacteriol, 2011, 193(10): 2460-2467. DOI:10.1128/JB.01307-10 |
| [6] |
梁文娟. 基于CRISPRs的大肠埃希菌分型方法及其与耐药和毒力关系[D]. 郑州: 郑州大学, 2017. Liang WJ. The genotyping method based on CRISPRS and the relationship between the CRISPR/Cas and the virulence and resistant in Escherichia coli[D]. Zhengzhou: Zhengzhou University, 2017. |
| [7] |
Shariat N, Dudley EG. CRISPRs: molecular signatures used for pathogen subtyping[J]. Appl Environ Microbiol, 2014, 80(2): 430-439. DOI:10.1128/AEM.02790-13 |
| [8] |
Joensen KG, Tetzschner AMM, Iguchi A, et al. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data[J]. J Clin Microbiol, 2015, 53(8): 2410-2426. DOI:10.1128/JCM.00008-15 |
| [9] |
Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria[J]. J Clin Microbiol, 2012, 50(4): 1355-1361. DOI:10.1128/JCM.06094-11 |
| [10] |
Zeng HY, Li CS, He WJ, et al. Cronobacter sakazakii, Cronobacter malonaticus, and Cronobacter dublinensis genotyping based on CRISPR locus diversity[J]. Front Microbiol, 2019, 10: 1989. DOI:10.3389/fmicb.2019.01989 |
| [11] |
Marraffini LA. CRISPR-Cas immunity in prokaryotes[J]. Nature, 2015, 526(7571): 55-61. DOI:10.1038/nature15386 |
| [12] |
刘璨颖, 张济培, 王丙云. 大肠杆菌O-抗原血清型鉴定研究进展[J]. 中国人兽共患病学报, 2016, 32(10): 928-933. Liu CY, Zhang JP, Wang BY. Research progress on identification of Escherichia coli O-antigen serogroups[J]. Chin J Zoon, 2016, 32(10): 928-933. DOI:10.3969/j.issn.1002-2694.2016.010.015 |
| [13] |
王龙光, 姜雯, 逄春华, 等. 利用MLST技术探讨不同地区致病性耐药鸡源大肠杆菌的遗传进化关系[J]. 中国兽医学报, 2017, 37(9): 1680-1686. Wang LG, Jiang W, Pang CH, et al. Genetic evolutionary relationship of drug-resistant and pathogenic chicken originated Escherichia coli strains from different regions with MLST[J]. Chin J Vet Sci, 2017, 37(9): 1680-1686. DOI:10.16303/j.cnki.1005-4545.2017.09.08 |
| [14] |
Yokoyama K, Makino K, Kubota Y, et al. Complete nucleotide sequence of the prophage VT1-Sakai carrying the Shiga toxin 1 genes of the enterohemorrhagic Escherichia coli O157: H7 strain derived from the Sakai outbreak[J]. Gene, 2000, 258(1-2): 127-139. DOI:10.1016/S0378-1119(00)00416-9 |
| [15] |
Kulasekara BR, Jacobs M, Zhou Y, et al. Analysis of the genome of the Escherichia coli O157:H7 2006 spinach- associated outbreak isolate indicates candidate genes that may enhance virulence[J]. Infect Immun, 2009, 77(9): 3713-3721. DOI:10.1128/IAI.00198-09 |
| [16] |
Ahmed SA, Awosika J, Baldwin C, et al. Genomic comparison of Escherichia coli O104:H4 isolates from 2009 and 2011 reveals plasmid, and prophage heterogeneity, including shiga toxin encoding phage stx2[J]. PLoS One, 2012, 7(11): e48228. DOI:10.1371/journal.pone.0048228 |
| [17] |
Ogura Y, Ooka T, Iguchi A, et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli[J]. Proc Natl Acad Sci USA, 2009, 106(42): 17939-17944. DOI:10.1073/pnas.0903585106 |
| [18] |
Cooper KK, Mandrell RE, Louie JW, et al. Comparative genomics of enterohemorrhagic Escherichia coli O145:H28 demonstrates a common evolutionary lineage with Escherichia coli O157:H7[J]. BMC Genomics, 2014, 15: 17. DOI:10.1186/1471-2164-15-17 |
| [19] |
Beatson SA, Ben Zakour NL, Totsika M, et al. Molecular analysis of asymptomatic bacteriuria Escherichia coli strain VR50 reveals adaptation to the urinary tract by gene acquisition[J]. Infect Immun, 2015, 83(5): 1749-1764. DOI:10.1128/IAI.02810-14 |
| [20] |
Stephens CM, Skerker JM, Sekhon MS, et al. Complete genome sequences of four Escherichia coli ST95 isolates from bloodstream infections[J]. Genome Announc, 2015, 3(6): e01241-15. DOI:10.1128/genomeA.01241-15 |
| [21] |
Yang SY, Liu J, Shao FY, et al. Analysis of the features of 45 identified CRISPR loci in 32 Staphylococcus aureus[J]. Biochem Biophys Res Commun, 2015, 464(3): 894-900. DOI:10.1016/j.bbrc.2015.07.062 |