 2022, Vol. 43
2022, Vol. 43文章信息
- 郭永豪, 豆巧华, 刘倩, 杨建辉, 吕宛玉, 丰达星, 僧明华, 张延炀, 赵东阳.
- Guo Yonghao, Dou Qiaohua, Liu Qian, Yang Jianhui, Lyu Wanyu, Feng Daxing, Seng Minghua, Zhang Yanyang, Zhao Dongyang
- 中国乙型肝炎病毒基因组序列突变及进化分析
- Analysis on the sequence mutation and evolution of HBV genome in China
- 中华流行病学杂志, 2022, 43(8): 1309-1314
- Chinese Journal of Epidemiology, 2022, 43(8): 1309-1314
- http://dx.doi.org/10.3760/cma.j.cn112338-20220411-00278
-
文章历史
收稿日期: 2022-04-11




HBV是一种小型包膜DNA病毒,属于肝病毒科。病毒基因组大约由3 200个核苷酸组成,有4个部分重叠的开放阅读框,分别为表面(S)蛋白、前核(Pre-core)/核(C)蛋白、聚合酶(P)和X蛋白。根据HBV全基因组或编码S蛋白基因序列的同源性,确定10个HBV基因型(A~J)和一些亚型[1]。在中国,B和C基因型HBV较为流行[2]。接种乙型肝炎(乙肝)疫苗可有效预防HBV感染,中国1992年将乙肝疫苗纳入计划免疫以来,HBV感染率、乙肝的发病率和死亡率显著下降[3]。随着疫苗接种率的提高,由于疫苗接种的免疫压力导致的免疫逃逸突变一直伴随着疫苗的接种而存在。HBV免疫逃逸突变主要发生于S蛋白主要亲水区,包括I/T126A/N/I/S、Q129H/R、M133L、K141E、P142S、D144A/E和G145R/A等位点突变[4]。虽然这些突变极少成为流行株,但是考虑到在全球很多国家和地区都分离到了免疫逃逸突变株,所以免疫逃逸突变依然可能对公共体系造成威胁[5-6]。用反转录酶核苷类似物抑制HBV治疗慢性感染患者,可抑制HBV复制并降低肝病进展的风险。然而,长期使用反转录酶抑制剂治疗会导致耐药性。拉米夫定最早用于治疗慢性乙肝,约14.32%的患者在第一年出现耐药性变异,治疗5年后,70%的患者可产生耐药突变[7],常见的拉米夫定耐药突变位点为rtM204I、rtM204V、rtA181V/T等[8]。为了更好地了解中国HBV的分子流行病学特征,本研究对中国1998-2021年提交的HBV序列进行分析,系统阐明免疫逃逸突变和耐药突变的分布特征,分析HBV演化的时钟频率。
材料与方法1. 研究设计和序列纳入标准:2022年2月16日利用NCBI基因序列数据库(www.ncbi.nlm.nih.gov/genbank/)对中国各省份提交的HBV全基因组序列进行搜索,检索条件为选择“Nucleotide”,然后输入检索关键词“HBV+China+省份名称”进行检索,为了减少非全序列的干扰,检索的基因组序列长度范围设置为3 100~3 300 bp。
2. HBV基因型分布、耐药及S基因突变的测定:使用在线工具(https://www.genafor.org/index.php)gen2pheno(HBV)检测HBV基因型、基因亚型、抗病毒药物耐药和S基因突变,并与GenBank数据进行比较,分析时,因为中国D亚型较少,未勾选内嵌的D4亚型序列作为参考序列。
3. HBV基因组的进化分析:使用BioEdit软件编辑基因组序列,使用MAFFT软件进行序列比对,使用Fasttree软件构建进化树并进行树编辑和优化,参数均使用默认参数。计算序列演化分析时,使用BEAST 1.10.4软件,选用分级似然比检验,选用最佳拟合核苷酸替代模型,蒙特卡罗-马尔科夫链取值为10 000 000,Burn-in取值为1 000 000,其他选用默认参数。
结果1. 纳入分析序列:共查到5 439条序列,有13条序列是来自蝙蝠,剔除,最后纳入分析5 426条。序列提交年份最早为1998年(AF100309.1),最晚为2021年(MZ439295)。
2.中国不同区域流行的主要毒株型别:共收集到19个省份提交的序列。提交序列最多的是北京市,共提交1 395条序列,占所有序列的25.7%(1 395/5 426)。其次为上海市,共提交1 306条序列,占所有序列的24.1%(1 306/5 426)。提交序列最少的为河南省,共提交2条序列(0.04%,2/5 426)。在提交的所有序列中,C型占比最高,共有3 211条序列(59.1%,3 211/5 426)。其次为B型,共有1 833条序列(33.7%,1 833/5 426),B型和C型合计占比为92.8%。D型共计233条序列(4.3%,233/5 426)。A型143条序列,G型有14条序列,H型有2条序列,A、G、H型共占2.9%(159/5 426)。B型和C型分布具有明显的地域特征,上海市、台湾地区、云南省、广东省、江西省的B型占比较高。其中台湾地区B型占比为89.5%(511/571),上海市的B型占比为39.3%(513/1 306),云南省B型占比为48.8%(363/744),广东省B型占比为55.0%(99/180)。C型占比最多的是香港地区(81.1%,86/106),其次是北京市(79.9%,1 115/1 395),广西壮族自治区次之(74.9%,434/578)。广西壮族自治区和云南省各自提交了7条和6条G型序列。
3. S蛋白的突变和免疫逃逸突变:S蛋白的突变具有典型的型别特征,A、B、C和D型的突变热点各不相同。在S蛋白突变的位点中,有一些突变发生于S蛋白的亲水区,会造成一定程度的免疫逃逸。在5 426条序列中,有764条序列发生了免疫逃逸突变(14.1%,764/5 426),A型S蛋白序列突变率最高的是第45位丝氨酸、第114位苏氨酸和第194位的丙氨酸,突变率均为89.5%,分别突变为丙氨酸、丝氨酸和缬氨酸。有38条序列(26.6%,38/143)发生了可能的免疫逃逸突变,最常见的突变位点为第134位氨基酸(22.4%)。B型突变频率最高的是第200位苯丙氨酸突变为酪氨酸,31.8%的序列发生了该位点的突变。B型有291条序列发生了免疫逃逸突变(15.9%,291/1 829),第129位氨基酸易发生突变(2.0%)。C型突变频率最高的是第3位的丝氨酸变为天冬酰胺,59.9%的序列发生了该位点的突变。有402条序列(12.5%,402/3 207)发生了免疫逃逸突变,最常见的突变位点为第126位氨基酸(2.6%)。D型的序列中,97.4%的序列第127位苏氨酸突变为脯氨酸。145位氨基酸免疫逃逸突变在B、C和D型中均有发现,C型最多,共计有96条序列(3.0%,96/3 207)发生了该免疫逃逸突变,其中突变为精氨酸(R)的占79条,突变为丙氨酸(A)的占17条。(表 1)。
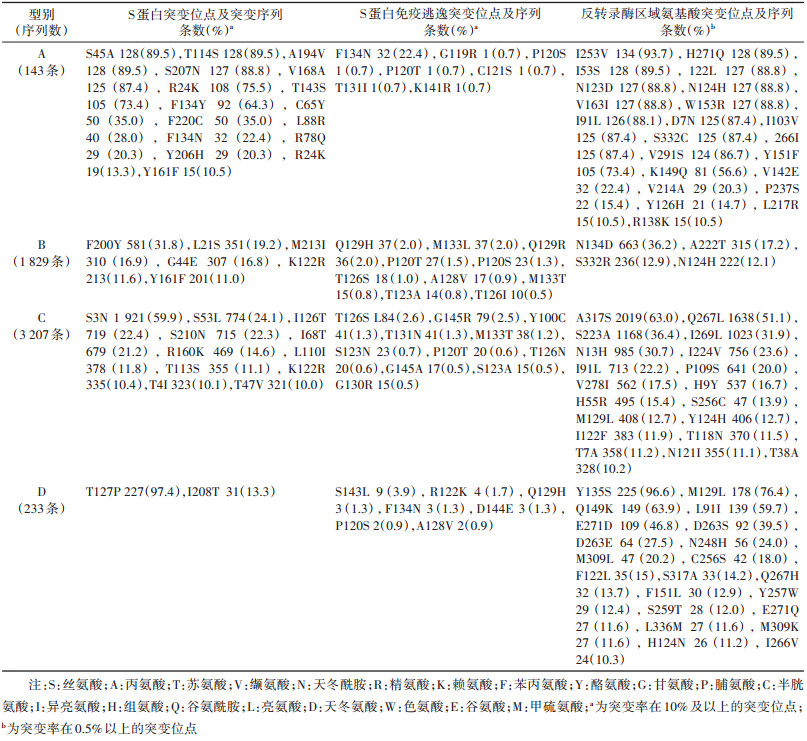
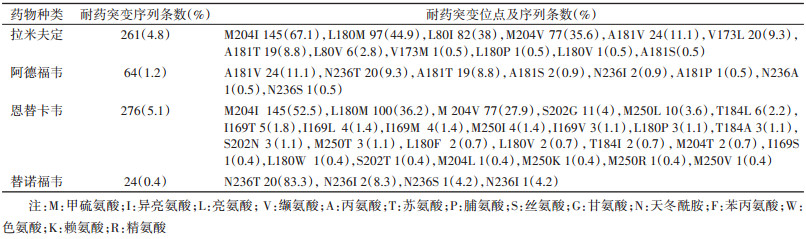
4. 耐药位点突变:HBV的耐药突变有耐一种药物突变,也可以发生耐多种药物突变。HBV的耐药位点突变主要发生于P蛋白的反转录酶氨基酸序列。5 426条序列中,只有101条序列未发生反转录酶蛋白编码序列突变,占1.9%(101/5 426),其余的98.1%的反转录酶蛋白序列至少发生了一处突变。在所有发生突变的序列中,共有399条序列发生了至少耐一种抗病毒药物(拉米夫定/恩替卡韦/替诺福韦/替比夫定)的耐药突变,占7.5%[399/(5 426-101)],总耐药突变率为7.4%(399/5 426)。
在4种抗病毒药物中,对恩替卡韦的耐药突变率最高(5.1%),其次为对拉米夫定的耐药突变(4.8%),最低的为替诺福韦(0.4%)。其中204位和180位氨基酸的突变会同时导致对拉米夫定和恩替卡韦的耐药,这两个位点突变率最高。在拉米夫定的耐药突变中204位氨基酸突变占67.1%,180位氨基酸突变占44.9%。在恩替卡韦耐药突变中,204位氨基酸突变占52.5%,180位氨基酸突变占36.2%。替诺福韦的耐药突变均发生于236位的氨基酸,分别突变为苏氨酸(T)、异亮氨酸(I)和丝氨酸(S)。见表 2。
5. HBV基因组进化分析:不同基因型分别位于不同的聚类分支。中国序列的C和B型处于两个主要分支上,其他型别处于B和C型之间,A、D、G和H型在进化关系上更近一些,处于同一个聚类分支上。将A、B、C和D型按照NCBI库中的ID号进行从A至M的排序,按照序列的总数量,C和B型按照1%的比例进行等间距抽样,A和D型按照1/30的比例进行等间距抽样,共计抽出66个序列用于HBV基因组进化分析。见图 1。与外部参考序列大猩猩的HBV毒株相比,选中分析的序列中,有3株来自于上海市(AF182802,AF100308,KY881792)、1株来自于西藏自治区(AY057948)和1株来自于陕西省(KC774379)的序列在距今3.5万~4.6万年之间出现了进化分支,其余序列均在距今6.5万年前出现了进化分支。进化的碱基替代速率为2.8113E-4(95%CI:1.101E-4~4.312E-4)。将外部参考序列(2株大猩猩HBV)去除后进行进化分析,以上提到的5株人感染HBV序列在5500年前出现了进化分支,多数中国人感染HBV的祖先序列均来自于公元1801年左右,进化的碱基替代速率为2.935E-4(95%CI:1.714E-4~4.068E-4)。

|
| 注:A型:紫色;B2亚型:深红色;B3亚型:淡黄色;B4亚型:粉红色;C1亚型:淡蓝色;C2亚型:蓝色;C4亚型:深蓝色;D型:绿色;G型和H型较少,未显示颜色 图 1 中国5 426条HBV基因组序列的聚类分析 |
HBV各种基因型的流行区域有较大的差别。A型主要分布于西欧、北美和非洲地区,B型和C型主要分布于包括中国在内的西亚太地区,D型分布于青藏高原、中亚地区和地中海周边国家。非洲部分地区有E型分布,南美洲部分地区有F型分布[9]。中国HBV基因型分布具有典型的地域特征,南部B型较多,大约占50%,北部地区C型占主导地位[10]。本研究中,中国提交的HBV全序列分析表明,C型占据着优势地位,其次为B型,二者合计占到了中国提交HBV全序列的92.8%。有研究表明,在HBV的各个基因型中,C型HBV慢性感染最易引起肝癌和肝硬化,其次为B型,两种基因型对于干扰素的抵抗作用高于其他基因型[11],这或许是中国HBV疾病负担较重的重要原因之一。中国香港地区提交的序列中,C型占比很高,与广东省有较大差别。在以前的文献中,也有报道中国香港地区的C型流行率高于B型[12],这或许与人口的迁徙相关。
B细胞抗原表位位于HBV S蛋白(主蛋白)的主要亲水区(major hydrophilic region,MHR;aa 99~169)。不同HBV的基因型有同样的抗原位点“a”(AA 124~147),为表面抗体(HBsAb)产生中和反应的特异性抗原位点,该区域氨基酸突变,可能会导致该抗原构象改变,从而导致抗原性的改变。中国提交的序列中,14.1%的序列发生了MHR区域的氨基酸突变,C型中,145位甘氨酸突变分布最广,其中突变为精氨酸(R)的占79条,突变为丙氨酸(A)的占17条。G145R是最早发现的HBV免疫逃逸突变,逃逸能力较强,90年代初,在一名接受乙肝疫苗和免疫球蛋白的意大利儿童中首次发现该突变[13]。Oon等[14]观察到G145R位置免疫逃逸突变在一段时间内是稳定的,也会发生水平传播。在我国之前的文献中也有报道[15]。因此,随着乙肝疫苗在全球更加广泛使用,应该更加关注疫苗接种可能引起的HBV免疫逃逸突变。
HBV的耐药突变一直是困扰临床治疗的难题,最经典也最常见的突变是P蛋白反转录酶区域180位的亮氨酸(L)204位的蛋氨酸(M)突变。204位常见突变为M突变为异亮氨酸(I)和缬氨酸(V)[16]。在使用拉米夫定治疗时,随着治疗时间的延长,耐药性不断延长。地中海沿岸国家的HBV序列中,有4.5%的序列发生了拉米夫定耐药突变,4.3%的序列发生了替比夫定耐药突变[17]。我国提交的HBV全基因组序列中,共有7.5%的序列发生了至少一处耐药突变,对拉米夫定的耐药突变率为4.8%,高于地中海沿岸国家的耐药突变率。
HBV的基因组可以插入到宿主的基因组内,具有较好的保守性,因此HBV的基因组进化比较缓慢。对一些800~4 500年前古代人尸体的HBV基因测序分析,计算出每个碱基每年的替换突变率为8.04×10-6~1.51×10-5,推测HBV进化的根应该在8 600年至2万年以前,现代人HBV的感染特征可能与新时期时代和青铜器时代人类的迁徙有关[18]。也有分析认为,HBV进化的根至少要追溯到1.2万年以前[19]。对中国2000-2006年的528条序列的研究表明,B型免疫逃逸的进化根在1997年以前,C型的进化根在1976年以前。本研究的基因组最早提交于1998年,最晚提交于2021年,时间跨度为34年。计算结果显示,以大猩猩的HBV序列作为外部参考序列分析显示,进化的根约在6.5万年以前,而中国本土主要流行的HBV序列进化的根约在200年前(公元1801年),有个别序列的根在5 000年以前。软件计算是一种基于碱基替代随机发生、突变不断积累的模型得出的结果,寻找HBV的起源时间还需要更多的医学证据,比如通过考古等方法,寻找石器时代人类的遗传物质,从而提供更有力的证据。本研究选用了NCBI数据库中收录的中国HBV数据,剔除了不是完整基因组序列的数据,因此在数据的选取上可能存在偏倚。
利益冲突 所有作者声明无利益冲突
作者贡献声明 郭永豪:文章撰写、数据分析、经费支持;豆巧华、刘倩、杨建辉:实验设计和数据分析;吕宛玉、丰达星、僧明华:统计学分析;张延炀、赵东阳:研究指导、论文修改
| [1] |
Kurbanov F, Tanaka Y, Mizokami M. Geographical and genetic diversity of the human hepatitis B virus[J]. Hepatol Res, 2010, 40(1): 14-30. DOI:10.1111/j.1872-034X.2009.00601.x |
| [2] |
Zeng G, Wang Z, Wen S, et al. Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotypes in China[J]. J Viral Hepat, 2005, 12(6): 609-617. DOI:10.1111/j.1365-2893.2005.00657.x |
| [3] |
Cui FQ, Shen LP, Li L, et al. Prevention of chronic Hepatitis B after 3 decades of escalating vaccination policy, China[J]. Emerg Infect Dis, 2017, 23(5): 765-772. DOI:10.3201/eid2305.161477 |
| [4] |
Lazarevic I, Banko A, Miljanovic D, et al. Immune-escape Hepatitis B virus mutations associated with viral reactivation upon immunosuppression[J]. Viruses, 2019, 11(9): 778. DOI:10.3390/v11090778 |
| [5] |
Koyaweda GW, Ongus JR, Machuka E, et al. Detection of circulating hepatitis B virus immune escape and polymerase mutants among HBV-positive patients attending Institut Pasteur de Bangui, Central African Republic[J]. Int J Infect Dis, 2020, 90: 138-144. DOI:10.1016/j.ijid.2019.10.039 |
| [6] |
Colagrossi L, Hermans LE, Salpini R, et al. Immune-escape mutations and stop-codons in HBsAg develop in a large proportion of patients with chronic HBV infection exposed to anti-HBV drugs in Europe[J]. BMC Infect Dis, 2018, 18(1): 251. DOI:10.1186/s12879-018-3161-2 |
| [7] |
Glebe D, Goldmann N, Lauber C, et al. HBV evolution and genetic variability: Impact on prevention, treatment and development of antivirals[J]. Antiviral Res, 2021, 186: 104973. DOI:10.1016/j.antiviral.2020.104973 |
| [8] |
Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances[J]. World J Gastroenterol, 2014, 20(24): 7653-7664. DOI:10.3748/wjg.v20.i24.7653 |
| [9] |
Lin CL, Kao JH. Hepatitis B virus genotypes and variants[J]. Cold Spring Harb Perspect Med, 2015, 5(5): a021436. DOI:10.1101/cshperspect.a021436 |
| [10] |
Zhu CT, Dong CL. Characteristics of general distribution of hepatitis B virus genotypes in China[J]. Hepatobiliary Pancreat Dis Int, 2009, 8(4): 397-401. |
| [11] |
Vincent W, Joseph J. Diagnosis and personalized management of hepatitis B including significance of genotypes[J]. Gastrointestinal Infections, 2012, 25(5): 570-577. |
| [12] |
Yuen MF, Sablon E, Tanaka Y, 等. 香港地区慢性乙型肝炎患者HBV基因型、核心启动子和前核心区变异的流行病学研究[J]. 世界核心医学期刊: 胃肠病学分册, 2005, 24(1): 61. Yuen MF, Sablon E, Tanaka Y, et al. Epidemiological study of HBV genotype, core promoter and pre-core region variation in patients with chronic hepatitis B in Hong Kong[J]. Digest World Core Med J: Gastroenterology, 2005, 24(1): 61. |
| [13] |
Zanetti AR, Tanzi E, Manzillo G, et al. Hepatitis B variant in Europe[J]. Lancet, 1988, 332(8620): 1132-1133. DOI:10.1016/S0140-6736(88)90541-7 |
| [14] |
Oon CJ, Chen WN, Goo KS, et al. Intra-familial evidence of horizontal transmission of hepatitis B virus surface antigen mutant G145R[J]. J Infect, 2000, 41(3): 260-264. DOI:10.1053/jinf.2000.0751 |
| [15] |
郭永豪, 丰达星, 董蒲梅, 等. 河南省乙型肝炎病毒分型特征及小S蛋白主要亲水区变异特点分析[J]. 病毒学报, 2017, 33(1): 44-48. Guo YH, Feng DX, Dong PM, et al. Analysis on genotype distribution and mutation of major hydroponic region of Hepatitis B Virus in Henan province[J]. Chin J Virol, 2017, 33(1): 44-48. DOI:10.13242/j.cnki.bingduxuebao.003098 |
| [16] |
Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues[J]. Gastroenterology, 2009, 137(5): 1593-1608.e2. DOI:10.1053/j.gastro.2009.08.063 |
| [17] |
Athamneh RY, Arıkan A, Sayan M, et al. Variable proportions of phylogenetic clustering and low levels of antiviral drug resistance among the major HBV sub-genotypes in the middle east and North Africa[J]. Pathogens, 2021, 10(10): 1333. DOI:10.3390/pathogens10101333 |
| [18] |
Ross ZP, Klunk J, Fornaciari G, et al. The paradox of HBV evolution as revealed from a 16th century mummy[J]. PLoS Pathog, 2018, 14(1): e1006750. DOI:10.1371/journal.ppat.1006750 |
| [19] |
Mühlemann B, Jones TC, Damgaard PDB, et al. Ancient hepatitis B viruses from the Bronze Age to the Medieval period[J]. Nature, 2018, 557(7705): 418-423. DOI:10.1038/s41586-018-0097-z |