 2021, Vol. 42
2021, Vol. 42文章信息
- 杨淞淳, 李重阳, 胡一祯, 孙秋芬, 潘建桥, 孙点剑一, 马宝山, 吕筠, 李立明.
- Yang Songchun, Li Chongyang, Hu Yizhen, Sun Qiufen, Pan Jianqiao, Sun Dianjianyi, Ma Baoshan, Lyu Jun, Li Liming
- gwasfilter:用于筛选全基因组关联研究的R脚本
- gwasfilter: an R script to filter genome-wide association study
- 中华流行病学杂志, 2021, 42(10): 1876-1881
- Chinese Journal of Epidemiology, 2021, 42(10): 1876-1881
- http://dx.doi.org/10.3760/cma.j.cn112338-20200731-01003
-
文章历史
收稿日期: 2020-07-31




2. 大连海事大学信息科学技术学院 116026;
3. 北京大学公众健康与重大疫情防控战略研究中心 100191
2. College of Information Science and Technology, Dalian Maritime University, Dalian 116026, China;
3. Peking University Center for Public Health and Epidemic Preparedness and Response, Beijing 100191, China
全基因组关联研究(genome-wide association study,GWAS)是在整个人类基因组范围内检验遗传变异(genetic variant)与疾病或性状之间统计学关联的重要研究方法[1]。深入分析GWAS发现的显著遗传位点对于确定疾病易感位点(casual variant)、阐明疾病遗传机制、实现精准医疗和精准预防都具有重要意义[2-5]。NHGRI-EBI GWAS Catalog(https://www.ebi.ac.uk/gwas/,GWAS Catalog)自2008年起持续收集所有已发表的符合该网站纳入标准的GWAS,是目前信息最为全面、更新相对及时、数据质量较高且能够公开获取的GWAS数据库[6]。截至2021年8月,GWAS Catalog已收录超过5 000篇GWAS文献的研究信息。
在报告新的GWAS位点(new loci)、构建遗传风险评分(genetic risk score)、开展基因多效性(pleiotropy)分析、遗传相关(genetic correlation)分析、孟德尔随机化(mendelian randomization)或基因-环境交互作用(gene-environment interaction)分析以及通径分析(pathway analysis)等之前[7-12],都需要检索、利用既往GWAS的研究结果。对已发表的GWAS研究通常要参考以下一些原则进行适当筛选,以获得可信度更高、与目标人群更相关的研究。例如,在GWAS中,发现人群中观察到的结果若能在另一个独立人群中得到验证(replication),则具有更高的可信度[13]。由于常见的遗传变异与疾病或性状之间的关联强度往往较弱,大样本量将更有助于发现潜在的关联[13]。同时,由于不同种族人群遗传背景的天然差异,在某个种族中发现的遗传位点可能无法直接应用于其他种族[13]。在GWAS Catalog中筛选目标GWAS研究时,应用上述这些条件,需要人工逐一判断和筛选,特别是同时需要筛选较多性状时,费时费力,且容易出错。有研究者最近开发了针对GWAS Catalog数据库的R程序包gwasrappid,提供的函数get_studies()只能基于特定的性状检索词查询GWAS Catalog中匹配的研究,但没有应用特定条件筛选研究的功能[14]。因此,我们开发了一套基于R语言的程序脚本,用于高效、准确地从GWAS Catalog中筛选满足特定条件的GWAS。
研究方法 (1) 筛选原理GWASCatalog网站允许直接下载完整的结构化数据库,包括研究设计、研究人群、性状和遗传位点等信息(图 1)。在GWAS Catalog中,性状主要采用Experimental Factor Ontology(EFO)分类系统,标准性状与EFO编码一一对应[6]。由于该分类系统具有跨域(cross-domain)分类的特点[15],通过某一EFO编码检索到的研究并非都能满足目标性状的要求。例如,若我们只关注在自然人群中开展的某性状的GWAS结果,则需剔除在患者中开展的GWAS、该性状对药物治疗反应的GWAS、该性状与其他性状交互作用的GWAS。因此,研究者首先需要根据GWAS Catalog提供的原始文献中“报告性状”(reported traits)的名称人工判断哪些是自己期望的性状,据此获取所有相关的GWAS后,则可以根据“是否有验证人群”“样本量大小”和“研究人群种族”等条件进一步筛选。

|
| 注:STUDY数据库中包含GWAS的发表、研究设计、性状等基本信息;ANCESTRY数据库中包含研究人群(含发现人群与验证人群)的种族及样本量信息;ASSOCIATION数据库中包含遗传位点与表型的关联信息及位点注释信息等。3个子数据库之间通过变量“STUDY ACCESSION”互相链接。数据库下载地址:https://www.ebi.ac.uk/gwas/docs/file-downloads 图 1 GWAS Catalog数据库基本结构 |
基于以上原理,由2名程序员采用R语言共同撰写了脚本gwasfilter.R[16]。撰写思路为:①将人工筛选研究的过程抽象为标准化的算法;②明确各环节的输入和输出;③采用R语言中的函数(function)对每个环节进行编译。该脚本基于R 3.5.1 x64环境开发,开发完成后,由3名测试员经过多次测试,在R 3.5软件及以上平台、32位操作系统及Linux操作系统均可正常运行。脚本源代码可在github平台(https://github.com/lab319/gwas_filter)
结果 (1) 脚本操作具体步骤gwasfilter.R包含6个标准步骤(图 2),内置5个预设函数(表 1),当前支持2种使用模式。模式1为从头筛选模式,包含完整的6个步骤,在未明确性状EFO编码和性状筛选词时使用。模式2为直接筛选模式,在已备好性状EFO编码文件和性状筛选词文件的前提下,可跳过步骤3~5直接进行步骤6。以下为各步骤简介:

|
| 图 2 gwasfilter.R脚本原理图 |
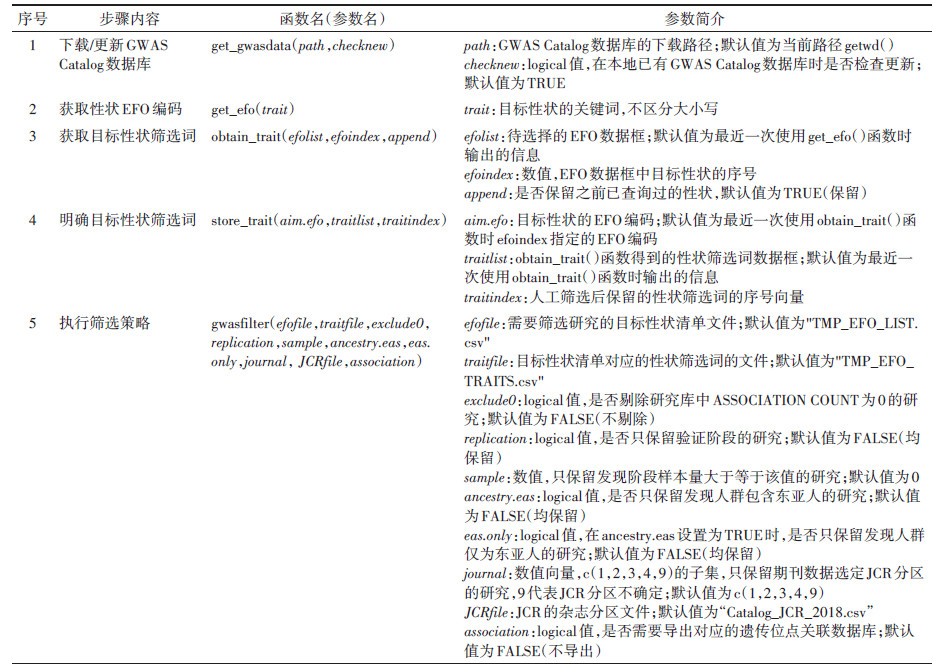
步骤1:准备R工作平台。在使用前需运用source命令加载gwasfilter.R中的预设函数。加载过程中程序会自动检查依赖包(readr、dplyr、stringr、data.table)的安装情况。
步骤2:下载/更新GWAS Catalog数据库。函数get_gwasdata()将GWAS Catalog网站的数据库下载至本地并读入内存。该函数会自动检查本地已有数据库是否需更新。
步骤3:获取目标性状EFO编码。函数get_efo()可返回查询给定关键词时所有相关的EFO编码。
步骤4:获取“报告性状”。函数obtain_trait()可返回在GWAS Catalog中检索某个EFO编码(一次只能选一个EFO编码)时所有相关研究的“报告性状”。
步骤5:明确目标性状筛选词。函数store_trait()通过直接记录正确的“报告性状”的序号向量traitindex,能方便地完成性状筛选词的存储过程。在执行步骤6前,步骤3~5可以循环进行(图 2),从而实现程序的批量筛选功能。
步骤6:执行筛选策略。函数gwasfilter()包含多个功能参数(表 1)。用户可以通过设置这些参数,灵活组合多个筛选条件:①是否剔除未观察到满足特定统计学显著性水平(P < 1.0×10-5)的遗传位点的研究(exclude0);②是否有验证人群(replication);③发现阶段样本量(sample);④发现人群的种族(ancestry.eas和eas.only);⑤期刊特征,如以2018年期刊引用报告(JCR)中的杂志分区(journal)为例。通过设置参数association=TRUE还可以额外导出入选研究对应的遗传位点关联数据库。在不设置任何筛选参数时,gwasfilter()可返回筛选前的所有记录。
(2) 测试情况以筛选总胆固醇水平(total cholesterol)的GWAS为例进行测试。测试所用平台为R 4.0.1软件,所用GWAS Catalog数据库的更新日期为2021-08-16,测试语句如下:
source("; gwasfilter.R”)#步骤1:加载脚本函数;
get_gwasdata()#步骤2:获取GWAS Catalog数据库;
get_efo(trait=“Cholesterol”)#步骤3:查询胆固醇相关的EFO编码;
obtain_trait(efoindex=1)#步骤4:查询目标性状对应的报告性状;
store_trait[traitindex=c(1,2,11,12)] #步骤5:明确目标性状筛选词;
gwasfilter(replication = T,ancestry.eas = T,eas.only = F,journal=c(1),association=T)#步骤6:执行筛选策略(有验证阶段,发现人群包含东亚人,期刊为JCR Q1区,同时输出对应的遗传位点关联数据库)。
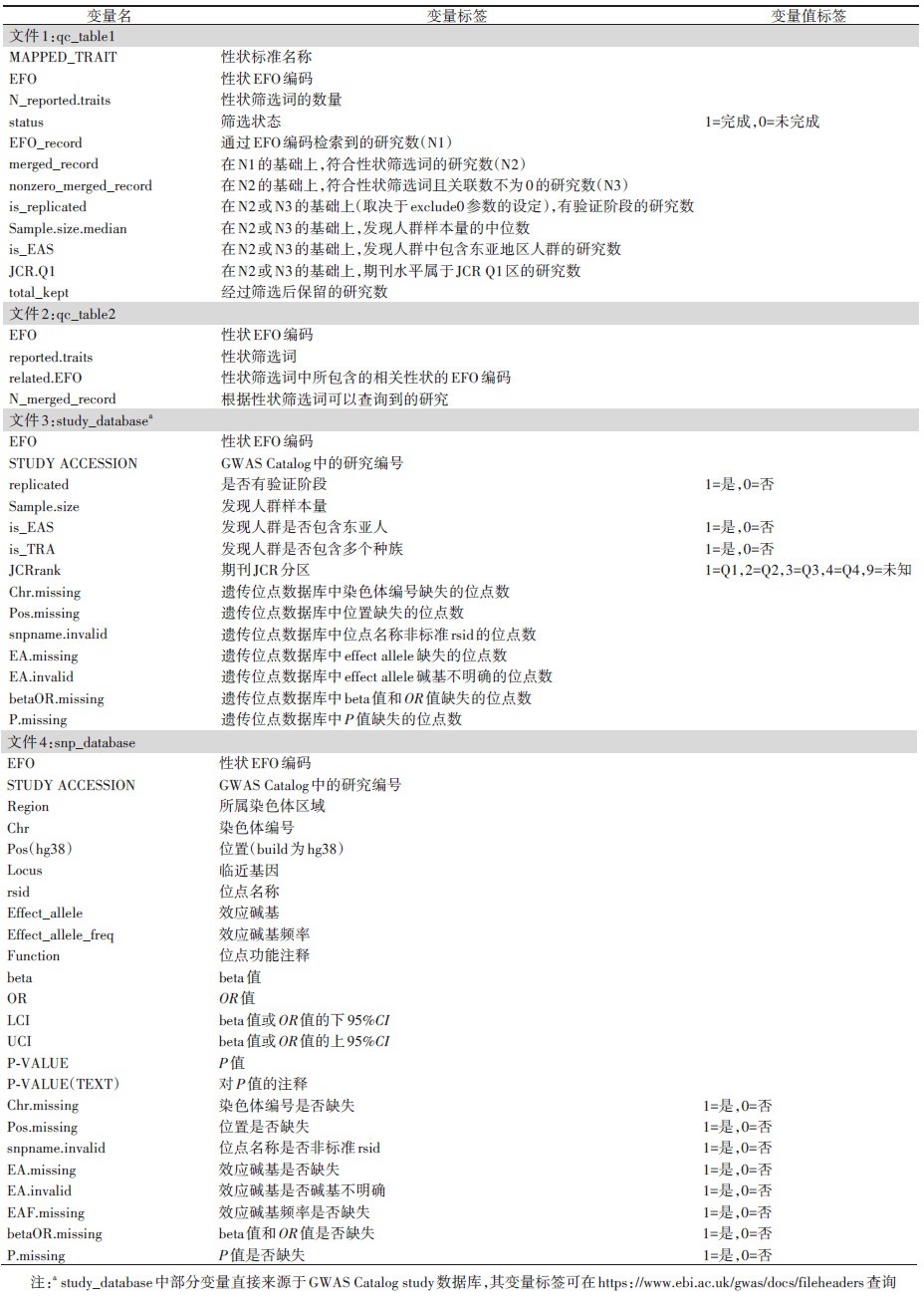
上述步骤中,除步骤2下载与预处理数据耗时约4 min以外(与具体网速有关),其余步骤程序用时均 < 1秒。步骤6完成后输出4个csv文件(表 2),分别为qc_table 1、qc_table 2、study_database和snp_database。在上述筛选条件下,共有6项研究符合筛选要求。
当前尚无其他的专注于筛选GWAS的工具,仅有的一个与GWAS Catalog相关的R程序包gwasrapidd仅能简单查询GWAS Catalog中的研究,没有进一步的筛选功能[14]。与人工筛选相比,gwasfilter.R具有4大优势。首先,使用过程简便。用户仅需要记录2个参数(efoindex和traitindex),并根据需要确定筛选策略,便可完成筛选,省去了在GWAS Catalog网站检索性状、下载数据然后逐一判断的繁琐过程;在筛选条件较多、筛选量较大时,也能避免人工操作导致的失误。第二,筛选过程高效。除下载数据外,程序执行过程所需时间几乎可以忽略。在需要筛选的性状较多时,脚本的批量筛选功能可以大幅度地提升筛选效率。第三,筛选过程标准化。gwasfilter.R导出的文件qc_table 1.csv记录了筛选过程的关键数据,每个性状所用的性状筛选词也能即时存储,便于对筛选过程进行核验;每个性状存储的性状筛选词也可在之后重复使用。第四,筛选方式灵活。用户可以根据研究需要,灵活地组合不同的筛选条件。
当然,gwasfilter.R也存在一定的局限性。第一,该脚本的运行依赖于GWAS Catalog定期发布的数据库,因此,筛选的准确度以及导出的遗传位点数据库的数据质量取决于GWAS Catalog数据库本身的数据质量。第二,在明确目标性状筛选词的环节(步骤5),不同研究者的意见可能存在分歧。通过多人平行地甄别性状筛选词将有助于提高筛选的准确度。第三,由于一项GWAS从发表到收录至GWAS Catalog有一定的时间间隔,对于3个月内新发表的研究,则需要在PubMed上检索。
综上所述,我们开发了一套基于GWAS Catalog数据库筛选GWAS的R脚本。该脚本操作简便,筛选研究的过程高效而标准化,可灵活应用于实际研究。后续研究可以在当前脚本的基础上进一步与其他公共数据库整合。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
陈峰, 柏建岭, 赵杨, 等. 全基因组关联研究中的统计分析方法[J]. 中华流行病学杂志, 2011, 32(4): 400-404. Chen F, Bai JL, Zhao Y, et al. Statistical methodologies used in genome-wide association studies[J]. Chin J Epidemiol, 2011, 32(4): 400-404. DOI:10.3760/cma.j.issn.0254-6450.2011.04.018 |
| [2] |
Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping[J]. Nat Rev Genet, 2018, 19(8): 491-504. DOI:10.1038/s41576-018-0016-z |
| [3] |
Gallagher MD, Chen-Plotkin AS. The post-GWAS era: from association to function[J]. Am J Human Genet, 2018, 102(5): 717-730. DOI:10.1016/j.ajhg.2018.04.002 |
| [4] |
Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores[J]. Nat Rev Genet, 2018, 19(9): 581-590. DOI:10.1038/s41576-018-0018-x |
| [5] |
Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications[J]. Nat Genet, 2015, 47(8): 856-860. DOI:10.1038/ng.3314 |
| [6] |
Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019[J]. Nucleic Acids Res, 2019, 47(D1): 1005-1012. DOI:10.1093/nar/gky1120 |
| [7] |
王铖, 戴俊程, 孙义民, 等. 遗传风险评分的原理与方法[J]. 中华流行病学杂志, 2015, 36(10): 1062-1064. Wang C, Dai JC, Sun YM, et al. Genetic risk score: principle, methods and application[J]. Chin J Epidemiol, 2015, 36(10): 1062-1064. DOI:10.3760/cma.j.issn.0254-6450.2015.10.005 |
| [8] |
Solovieff N, Cotsapas C, Lee PH, et al. Pleiotropy in complex traits: challenges and strategies[J]. Nat Rev Genet, 2013, 14(7): 483-495. DOI:10.1038/nrg3461 |
| [9] |
Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits[J]. Nat Genet, 2015, 47(11): 1236-1241. DOI:10.1038/ng.3406 |
| [10] |
林丽娟, 魏永越, 张汝阳, 等. 孟德尔随机化方法在观察性研究因果推断中的应用[J]. 中华预防医学杂志, 2019, 53(6): 619-624. Lin LJ, Wei YY, Zhang RY, et al. Application of mendelian randomization methods in causal inference of observational study[J]. Chin J Prev Med, 2019, 53(6): 619-624. DOI:10.3760/cma.j.issn.0253-9624.2019.06.015 |
| [11] |
McAllister K, Mechanic LE, Amos C, et al. Current challenges and new opportunities for gene-environment interaction studies of complex diseases[J]. Am J Epidemiol, 2017, 186(7): 753-761. DOI:10.1093/aje/kwx227 |
| [12] |
Wang K, Li MY, Hakonarson H. Analysing biological pathways in genome-wide association studies[J]. Nat Rev Genet, 2010, 11(12): 843-854. DOI:10.1038/nrg2884 |
| [13] |
McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges[J]. Nat Rev Genet, 2008, 9(5): 356-369. DOI:10.1038/nrg2344 |
| [14] |
Magno R, Maia AT. gwasrapidd: an R package to query, download and wrangle GWAS catalog data[J]. Bioinformatics, 2019, 36(2): 649-650. DOI:10.1093/bioinformatics/btz605 |
| [15] |
Malone J, Holloway E, Adamusiak T, et al. Modeling sample variables with an experimental factor ontology[J]. Bioinformatics, 2010, 26(8): 1112-1118. DOI:10.1093/bioinformatics/btq099 |
| [16] |
R Core Team. R: A language and environment for statistical computing[EB/OL]. R Foundation for Statistical Computing, Vienna, Austria, 2018. https://www.R-project.org/.
|