 2020, Vol. 41
2020, Vol. 41文章信息
- 蔡亮, 张恒娇, 何方玲, 奉雅乐, 胡世雄, 王娟, 刘富强, 蒋永林, 谭夏林, 潘海明, 唐宾彬, 杨浩, 龙浩宇, 湛志飞, 高立冬.
- Cai Liang, Zhang Hengjiao, He Fangling, Feng Yale, Hu Shixiong, Wang Juan, Liu Fuqiang, Jiang Yonglin, Tan Xialin, Pan Haiming, Tang Binbin, Yang Hao, Long Haoyu, Zhan Zhifei, Gao Lidong
- 湖南省2018年登革热本地暴发流行病学和病毒分子特征分析
- Epidemiological and virus molecular characterization of dengue fever outbreak in Hunan province, 2018
- 中华流行病学杂志, 2020, 41(12): 2119-2124
- Chinese Journal of Epidemiology, 2020, 41(12): 2119-2124
- http://dx.doi.org/10.3760/cma.j.cn112338-20200107-00018
-
文章历史
收稿日期: 2020-01-07




2. 永州市祁阳县疾病预防控制中心, 祁阳 426100;
3. 永州市疾病预防控制中心 425000
2. Qiyang Country Center for Disease Control and Prevention of Yongzhou, Qiyang 426100, China;
3. Yongzhou Prefectural Center for Disease Control and Prevention, Yongzhou 425000, China
登革热是由黄病毒科的登革病毒1、2、3和4型血清型引起的急性蚊媒传染病,主要通过埃及伊蚊和白纹伊蚊叮咬传播,是世界上分布广、发病率高、危害大的一种虫媒病毒感染性疾病[1-3],据WHO统计,目前全球有128个国家或地区,约39亿人口处于登革热感染的风险,年发病数3.9亿人[4]。人感染后可呈现无症状的隐性感染、登革热、登革出血热以及登革休克综合征[5-6]。2018年以前,湖南省的登革热疫情主要以散发的输入性病例为主。2018年9月上旬,湖南省永州市祁阳县某医疗机构报告当地相继出现发热、乏力、白细胞减少、血小板减少症状患者,为核实引起疾病的原因,本研究对病例标本开展了血清学、病原学和分子生物学的检测,确定为登革病毒感染后,采取了及时有效的防控措施,加强了病例监测。
材料与方法1.细胞株与试剂:C6/36细胞来源于中国CDC病毒病所脑炎室,MEM(Gibco,Carlsbad,USA),小牛血清(Gibco,Carlsbad,USA),登革病毒IgM、IgG抗体检测试剂盒(Capture ELISA,Panbio,Australian),NS1抗原检测试剂盒(胶体金法,Cortez,USA),病毒核酸提取试剂盒(磁珠法,西安天隆科技有限公司),登革病毒核酸检测试剂盒(Real-time PCR法,硕世生物,中国)。
2.病例发现和病原学诊断:2018年9月11日,湖南省永州市CDC接到冷水滩区CDC报告,永州市中心医院集中收治了9例来自祁阳县龙山街道的不明原因发热、全身乏力等不适症状病例,临床表现均有发热、白细胞和血小板减少,其中4例有皮疹、7例有胃肠道症状、2例ALT轻度增高。冷水滩区CDC对其中8例病例开展流行病学调查和血液标本采集,经市CDC送省CDC开展可疑病原体筛查,省CDC应用Real-time PCR及血清学方法对8份病例标本开展肠道病毒、新型布尼亚病毒、汉坦病毒、登革病毒的核酸、抗原、抗体检测。
3.病例搜索和实验室检测:病例确诊后,在祁阳县宝塔街社区及周边社区开展疑似登革热病例搜索和调查,重点采集近期有发热、头疼、眼眶疼、皮疹及出血症状的疑似病例以及与8例病例有密切接触的居民血清,依据《登革热诊断标准》(WS 216-2008)对标本开展登革病毒核酸、NS1抗原及病毒分离。
4. RNA提取、E基因扩增与序列测定:选取15株有代表性病例血清病毒分离培养物,磁珠法提取病毒核酸,应用5′ -GTGTCCCAGCCGGCGGTGTC ATCAGC-3′作为反转录引物构建病毒cDNA文库,应用登革热2型特异性引物扩增病毒E基因,对扩增产物应用BigDyeTM Terminator 3.1 Ready Reaction Mix进行扩增和序列测定(3730序列分析仪)。
5.序列比对和进化分析:应用PubMed-Entrez Search从GenBank、EMBL数据库中检索登革热2型E基因序列,下载44个不同基因型代表株的E蛋白全基因序列,其中全球型12株、美亚型4株、亚洲1型9株、亚洲2型6株、美洲型11株、森林型2株,建立数据库,应用Seqman、ClustalX2、Bioedit等软件计算核苷酸和氨基酸序列的相似度和变异度,Mege 6.0软件构建病毒E基因序列进化树(Neighbor-Joining法),分析可能的传播来源。
6.蚊媒监测:采用布雷图指数法在5-10月期间对永州市蚊媒密度开展日常监测,疫情发生后选择祁阳县浯溪街道王府坪社区、长虹街道人民西路社区、龙山街道邵家岭社区采用布雷图指数法开展蚊媒密度应急监测,计算风险指数,评估传播风险。
7.人群抗体水平监测:2019年6月,采取回顾性调查方式,在祁阳县随机采集377名健康人群血清,应用ELISA(捕获法)检测登革热IgG抗体,评估健康人群抗体水平。
8.统计学分析:采用回顾性流行病学与描述性流行病学方法,对疫情资料与监测数据进行统计分析,计算发病率和死亡率,在时间、空间和人群分布上分析登革热本地暴发流行的特点。统计分析采用SPSS 18.0软件进行,采用χ2检验比较不同率之间的差异,以P<0.05为差异有统计学意义。
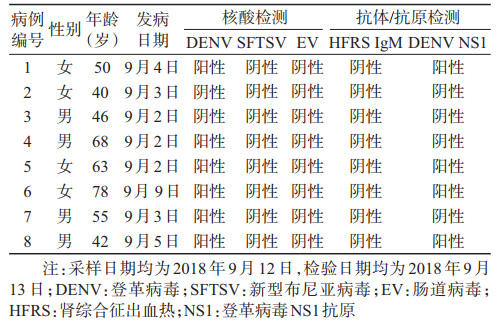
结果1.指示病例实验室确诊:对8例疑似病例血清开展登革病毒核酸、登革病毒NS1抗原、肠道病毒核酸、新型布尼亚病毒核酸检测,结果显示6例病例登革病毒核酸阳性,均居住在祁阳县龙山街道宝塔街社区,最早发病时间为9月2日,其中有4例病例登革病毒NS1抗原阳性。经病毒血清型分型,6例登革病毒核酸阳性标本均为登革病毒2型。8例病例标本肠道病毒和新型布尼亚病毒核酸均为阴性、汉坦病毒IgM和IgG抗体均为阴性。组织专家讨论认定,该起疫情为本地登革热感染暴发疫情,达到突发公共卫生事件标准,当日在突发公共卫生事件管理信息系统进行网络直报并派防控专家前往现场指导疫情处置和开展病例搜索。见表 1。
2.流行病学特征:累计搜索、检测、排查疑似登革热病例186例,确诊96例;发病年龄最小10岁,最大86岁;男性47例,女性49例,包括实验室确诊82例,临床诊断14例。重症病例6例,均伴有基础性疾病,临床表现以较严重出血为主,其中1例出现休克。轻症病例主要临床表现以发热(91/96)、乏力(73/96)、酸痛(47/96)为主,部分病例面、颈、胸发红,头疼、眼眶痛等症状。96例登革热感染暴发疫情集中发生在永州市祁阳县,波及2个乡镇的3个街道的17个社区/村,均由感染登革病毒2型所致。最早病例发病日期是9月1日,最晚发病日期是10月19日,疫情持续时间49 d,当地日平均气温28.16 ℃。综合分析发病时间、发病人数和当日最高气温,病例集中分布在9月13日前后,随着防控措施的实施和气温的逐渐降低,病例发生逐渐减少,发病曲线符合疾病流行模式,自9月15日后,峰值逐渐降低。见图 1。

|
| 图 1 湖南省2018年登革热本地暴发流行96例确诊病例发病日期与日最高气温之间的关系 |
3.病例核酸检测、病毒分离和基因测序:对96例确诊病例血清开展核酸检测、NS1抗原检测及病毒分离,核酸检测阳性72例(75.00%)、NS1抗原检测阳性61例(63.54%)、C6/36细胞分离到病毒株64株(66.67%),全部为登革病毒2型。选择不同发病地点、年龄、时间的15株毒株进行病毒E基因序列测定,获得15个登革病毒2型E基因全序列,基因全长1 485 bp,双链分子量901.45×106,AT含量55.22%,GC含量44.78%,将序列上传至GenBank获得的序列号为:MK931408、MK931409、MK931410、MK931411、MK931412、MK931413、MK931414、MK931415、MK931416、MK931417、MK931418、MK931419、MK931420、MK931421、MK931422。氨基酸位点同源性分析表明,15株毒株E蛋白氨基酸同源性达99.7%~100.0%;与全球型的浙江杭州株同源性最高,达99.4%~99.6%,与全球型的广东株同源性达96.3%~96.5%,与全球型其他代表株有一定差异,在93.6%~97.0%之间。E蛋白重要氨基酸位点分析表明,分离的毒株所有抗原位点E322均突变成V。
4.蚊媒密度监测:日常蚊媒密度监测提示,永州市2018年5-10月布雷图指数分别为4.84、8.74、18.50、10.92、22.22、2.27,6个月平均指数为11.29。疫情发生后祁阳县浯溪街道王府坪社区、长虹街道人民西路社区和龙山街道邵家岭社区蚊媒密度较高,布雷图指数分别为16、65、21,提示该区域存在极高的登革热流行风险,立即动员社区力量,采取灭蚊和滋生地的清理措施。
5.健康人群抗体监测:随机抽样的377名健康人群,男性142名,女性235名,其中1~岁组28名、20~岁组51名、40~岁组150名、≥60岁组148名。登革病毒IgG抗体阳性2例,抗体阳性率0.53%(2/377),均来自2018年疫情发生社区,2例病例均为女性,均>60岁,近期均无外出史。
6.亲缘进化分析:15株病毒(MK931408~ MK931422)与登革病毒2型的全球型毒株序列处于同一分支,属于登革病毒2型的全球型,与登革病毒2型的美亚型、亚洲1型、亚洲2型、美洲型、森林型进化距离较远,说明本次疫情仅由单一的登革病毒2型全球亚型引起。15株病毒与2017年广州株GD17040株、GZ217XN21274株(MH827554、MK564481)、2017年杭州株HZ-74株、HZ-49、HZ-1560、HZ-1003株(MH110719、MH010605、MH110612、MH110564)序列聚集在一起,亲缘关系较近,而与其他地区的登革毒株序列明显相距较远。见图 2。

|
| 图 2 2018年湖南省登革热本地暴发流行15株分离病毒株与其他代表毒株E基因进化分析 |
登革热在我国首次暴发流行发生在1978年的广东省,由登革病毒4型感染引起,造成22 122人感染,14人死亡,随后疫情逐渐向海南省、广西壮族自治区、福建省、浙江省和云南省蔓延[7]。湖南省自2005年起建立了包括输入性和本地疑似病例监测、传播媒介监测和人群血清学监测体系,近15年来每年均有输入性的病例报告,2018年由输入性登革热病例引起本地暴发流行是自监测以来的重要发现,全年累计报告登革热病例175例,报告病例数较2017年(26例)上升573.07%,其中本地感染病例96例,输入性病例79例,发病率0.25/10万,死亡1例,实验室确诊146例,临床诊断29例,输入性病例主要来自泰国、柬埔寨、缅甸、马来西亚、菲律宾和老挝及我国广东省。
祁阳县位于湖南省西南部,湘江中上游,属亚热带季风湿润气候,常住人口87.99万,登记的流动人口20余万,外出务工、经商人口较多,经济较发达,与广东、浙江省等地人员、贸易往来频繁。本次疫情中发现的6例指示病例发病前2周内均无外出史,可以判定为输入性病例引起的本地暴发疫情,在启动突发公共卫生事件应急响应后,通过在社区和医疗机构搜索病例,确诊登革热感染病例96例,流行病学调查显示96例病例发病前14 d均未曾离开过本地,说明为二代或三代病例。疫情发生后,及时采取了隔离治疗患者、灭成蚊、清除蚊虫滋生地、健康教育等措施,阻止了疫情向周边县(区)的进一步扩散。由于及时的灭蚊,未能对疫点蚊虫开展检测,目前蚊虫携带病毒的情况不明。
由于缺少有效的登革热疫苗和特效的治疗药物,人群对登革病毒普遍易感[8],2008-2016年湖南省健康人群IgG抗体阳性率达6.71%[9],说明存在既往或隐性感染病例。本研究通过回顾性地对祁阳县377名健康人群开展血清流行病学调查,IgG抗体阳性率为0.53%,2例病例均为祁阳县龙山街道和宝塔街道居民,近期无外出史,推测可能为2018年暴发疫情中的隐形感染病例。
登革病毒2型有美洲型、美亚型、亚洲1型、亚洲2型、森林型、全球型6个基因亚型[10-11],E基因介导了病毒和细胞受体的结合,与病毒的组织亲嗜性有关,是研究登革病毒分子流行病学的最适基因,适合分析暴发流行的毒株来源和生物学特性[12-13]。本研究对15株病毒分离株进行E基因全序列测序与分析,结果显示引起本次登革热暴发流行的毒株均为登革病毒2型全球型,与2017年广州株、2017年杭州株亲缘关系最近,推测引起本次暴发流行的毒株源自广东或浙江省,属于输入性病毒引起的本地暴发流行,广州市2001-2015年流行的DENV-2也主要以全球型为主[14],与流行病学调查资料一致。登革病毒2型毒株E蛋白主要毒力位点为E71(D)、E126(K)、E203(N)、E383~385(E-P-G)和E390(H)[15],15株毒株均存在抗原位点E322突变成V,关键性的毒力位点没有发生改变,其生物学意义有待进一步研究。
在暴发期间对发热病例血清开展病毒分离是为确诊登革热病例提供病原学证据的最可靠方法,本研究中登革病毒分离阳性率为66.67%,这是湖南省首次从病例血清分离到登革病毒,另有32例病例标本未能分离到病毒,结合核酸和NS1抗原检测结果分析,主要与标本采集时大部分病例过了病毒血症期有关。登革热病例在不同的病程,采用不同的检测方法检出阳性率不一致,本研究对96例病例血清应用PCR检测核酸、胶体金法检测NS1抗原、C3/36分离病毒,阳性率分别为75.00%、63.54%、66.67%,3种检测方法比较无显著性差异,但单一的方法检测均存在漏检,对登革热病例实验室确诊,必须多种方法联合运用,综合判断。
湖南省每年均有输入性的病例报告,此次暴发的原因,主要与全球化进程进一步扩大,湖南省同登革热流行地区的旅游、贸易、劳务日益频繁有关,增加了输入性病例的风险,同时湖南省8-10月间持续高热潮湿,14个市(自治州)均存在白纹伊蚊的滋生地,气温升高和降水增多,增加了蚊虫的滋生和活动能力,日常蚊媒密度监测永州市布雷图指数达11.29,具有较高的登革热传播风险,疫情发生后本地病例最多的3个社区布雷图指数高达16、65、21,具有极高的登革热传播风险。其次,与湖南省除了散发的输入性病例外,从未发生过本地流行,大部分病例为轻症或隐性感染,临床医生对该病的诊断能力欠缺,限制了病例的发现和报告有关。
综上所述,现场流行病学调查和分子遗传分析提示,湖南省2018年本地暴发疫情由输入性病例引起,由单一的登革病毒2型全球型引起。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
Gubler DJ. Epidemic Dengue/Dengue Hemorrhagic fever as a public health, social and economic problem in the 21st century[J]. Trends Microbiol, 2002, 10(2): 100-103. DOI:10.1016/S0966-842X(01)02288-0 |
| [2] |
Halstead SB. Dengue[J]. Lancet, 2007, 370(9599): 1644-1652. DOI:10.1016/S0140-6736(07)61687-0 |
| [3] |
San Martin JL, Brathwaite O, Zambrano B, et al. The epidemiology of dengue in the Americas over the last three decades:a worrisome reality[J]. Am J Trop Med Hyg, 2010, 82(1): 128-135. DOI:10.4269/ajtmh.2010.09-0346 |
| [4] |
Brady OJ, Gething PW, Bhatt S, et al. Refining the global spatial limits of dengue virus transmission by evidence-based consensus[J]. PLoS Negl Trop Dis, 2012, 6(8): e1760. DOI:10.1371/journal.pntd.0001760 |
| [5] |
Cruz CD, Forshey BM, Juarez DS, et al. Molecular epidemiology of American/Asian genotype DENV-2 in Peru[J]. Infect Genet Evol, 2013, 18: 220-228. DOI:10.1016/j.meegid.2013.04.029 |
| [6] |
Messina JP, Brady OJ, Scott TW, et al. Global spread of dengue virus types:mapping the 70 years history[J]. Trends Microbiol, 2014, 22(3): 138-146. DOI:10.1016/j.tim.2013.12.011 |
| [7] |
Li G, Pan P, He QY, et al. Molecular epidemiology demonstrates that imported and local strains circulated during the 2014 dengue outbreak in Guangzhou, China[J]. Virol Sin, 2017, 32(1): 63-72. DOI:10.1007/s12250-016-3872-8 |
| [8] |
W HO. Dengue:Guidelines for Diagnosis, Treatment, Prevention and Control[J]. Geneva:World Health Organization, 2009. DOI:10.1001/jama.1924.02650380058035 |
| [9] |
蔡亮, 孙倩莱, 何方玲, 等. 2008-2016年湖南省登革热病例及病原学监测结果[J]. 热带医学杂志, 2019, 19(11): 1425-1430, 1441. Cai L, Sun QL, He FL, et al. Epidemiology and virological characteristics of the dengue fever surveillance in Hunan province from 2008 to 2016[J]. J Trop Med, 2019, 19(11): 1425-1430, 1441. DOI:10.3969/j.issn.1672-3619.2019.11.026 |
| [10] |
Sang SW, Chen B, Wu HX, et al. Dengue is still an imported disease in China:a case study in Guangzhou[J]. Infect Genet Evol, 2015, 32: 178-190. DOI:10.1016/j.meegid.2015.03.005 |
| [11] |
Mir D, Romero H, de Carvalho LMF, et al. Spatiotemporal dynamics of DENV-2 Asian-American genotype lineages in the Americas[J]. PLoS One, 2014, 9(6): e98519. DOI:10.1371/journal.pone.0098519 |
| [12] |
Drumond BP, Mondini A, Schmidt DJ, et al. Population dynamics of DENV-1 genotype V in Brazil is characterized by co-circulation and strain/lineage replacement[J]. Arch Virol, 2012, 157(11): 2061-2073. DOI:10.1007/s00705-012-1393-9 |
| [13] |
Rico-Hesse R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature[J]. Virology, 1990, 174(2): 479-493. DOI:10.1016/0042-6822(90)90102-W |
| [14] |
刘远, 蒋力云, 罗雷, 等. 广州市2001-2015年登革病毒2型E基因进化分析[J]. 中华流行病学杂志, 2017, 38(1): 90-95. Liu Y, Jiang LY, Luo L, et al. Phylogenetic analysis of envelope gene of dengue virus serotype 2 in Guangzhou, 2001-2015[J]. Chin J Epidemiol, 2017, 38(1): 90-95. DOI:10.3760/cma.j.issn.0254-6450.2017.01.018 |
| [15] |
Lewis JA, Chang GJ, Lanciotti RS, et al. Phylogenetic relationships of dengue-2 viruses[J]. Virology, 1993, 197(1): 216-224. DOI:10.1006/viro.1993.1582 |