 2020, Vol. 41
2020, Vol. 41文章信息
- 李保娣, 何军, 李红育, 张慧, 徐丛杉, 于德山, 李建兵, 何健.
- Li Baodi, He Jun, Li Hongyu, Zhang Hui, Xu Congshan, Yu Deshan, Li Jianbing, He Jian
- 甘肃省人感染和外环境来源的H9N2禽流感病毒基因特征分析
- Analysis on genetic characteristics of H9N2 avian influenza virus isolated from human infection and external environment in Gansu province
- 中华流行病学杂志, 2020, 41(8): 1345-1351
- Chinese Journal of Epidemiology, 2020, 41(8): 1345-1351
- http://dx.doi.org/10.3760/cma.j.cn112338-20190712-00518
-
文章历史
收稿日期: 2019-07-12




2. 安徽省疾病预防控制中心, 合肥 230601;
3. 嘉峪关市疾病预防控制中心 735100
2. Anhui Provincial Center for Disease Control and Prevention, Hefei 230601, China;
3. Jiayuguan Center for Disease Control and Prevention, Jiayuguan 735100, China
H9N2禽流感病毒在世界范围内禽类中广泛流行,同时也能感染人,被认为具有大流行的潜力,具有重要的公共卫生学意义[1]。根据遗传学和流行病学将H9N2禽流感病毒分为北美和欧亚两大谱系[2]。欧亚谱系进一步进化分成3个主要分支:A/chicken/Beijing/1/1994(BJ/94-like)、A/quail/Hong Kong/G1/1997(G1-like)和A/duck/Hong Kong/Y439/1997(Y439-like)[2]。1998年出现一个新的支系A/chicken/Shanghai/F/1998(F/98-like)引起我国东部地区鸡群暴发流行,之后F/98-like与G1-like等重配为A/chicken/Human/5260/2005-like[3]。BJ/94和G1类似株在1990年后分别成为我国北方地区和南方地区主要禽类流行株。与中亚和中东流行的H9N2禽流感病毒相比,中国分离株是从HA和NA基因的系统发育树中独立聚类的[4]。
通过全球流感基因共享数据库(GISAID)搜索,目前全球已上传38例人感染H9N2禽流感病毒病例基因组序列:分布在阿曼、巴基斯坦、孟加拉国和韩国等国家,我国报道的人感染H9N2禽流感病毒病例主要分布在北京市、广东省、广西壮族自治区、安徽省、江西省、湖南省、四川省和中国香港地区。2016年8月甘肃省嘉峪关市发现1例人感染H9N2禽流感病毒病例,经治疗后痊愈。经甘肃省CDC流感参比中心确诊,后送中国CDC病毒病预防控制所国家流感中心进一步分离培养,获得A/Gansu_jiayuguan/1397/2016(H9N2)(GS_JYG/1397)毒株1株。全基因组序列分析表明为系间多次重配株。本研究分析甘肃省人感染和外环境来源的H9N2流感病毒序列数据,与我国相关的病毒序列数据进行比对,描述H9N2病毒全基因组分子进化特征,分析其重要氨基酸位点突变情况,探讨人感染H9N2禽流感病毒的来源,为人感染H9N2禽流感病毒防控提供依据。
材料与方法1.样本和数据来源:样本来自嘉峪关市的国家级流感监测哨点医院,由嘉峪关市CDC流感监测网络实验室负责样本初步鉴定,如果为H9亚型阳性,该标本冷链运输至甘肃省CDC流感参比中心及中国CDC病毒病预防控制所国家流感中心进行复核鉴定和基因测序,确认为人感染H9N2禽流感病毒病例。外环境监测在甘肃省14个市(州)各监测点采集禽类粪便、饮水及笼具内表面涂抹物等,于4 ℃条件24 h之内送到当地CDC国家级流感监测网络实验室进行病毒核酸检测,并将标本信息及时录入禽流感监测系统。阳性标本送甘肃省CDC参比中心复核鉴定,并送国家流感中心进一步分离培养及序列测定。禽类H9N2全基因组数据从美国国家生物技术信息中心(NCBI)和GISAID下载。
2.核酸检测[5]:对病例咽拭子样本进行荧光RT-PCR检测,使用QIAGEN公司的Rneasy MiNi Kit(货号:74104)试剂盒进行核酸提取,ThermoFisher公司的AgPath-IDTM One-step RT-PCR Reagents试剂盒(货号:AM1005)做为反应体系,并在ThermoFisher公司的7500荧光PCR仪上进行反应扩增。检测A型通用、A(H1N1)pdm09、H3、H5、H7、H9、N1、N6、N9亚型。所有引物和探针均由国家流感中心提供,所有实验操作均按照试剂盒说明书进行。
3.病毒分离培养:将H9N2禽流感病毒核酸检测阳性标本经无菌处理后接种于MDCK细胞分离培养流感病毒。然后用血凝和血凝抑制法鉴定流感病毒的型别/亚型。代表株抗原及参考血清为国家流感中心提供。
4. PCR产物纯化与测序:所有阳性标本和毒株送国家流感中心进行全基因组测序。
5.序列来源及参比序列:该病例所获得的H9N2毒株全基因组序列信息从国家流感中心病毒基因序列数据库中获取;参考序列选择A/turkey/Wisconsin/1/1966(H9N2)-like(TY/W1/66)、A/chicken/ Beijing/1/1994(H9N2)-like(BJ/94)、A/quail/Hong Kong/G1/1997(H9N2)-like(G1/97)、A/chicken/Hong Kong/G9/1997(H9N2)-like(G9/97)、A/duck/Hong Kong/Y439/1997(H9N2)-like(Y439/97)、A/duck/Hong Kong/d73/1976(H6N1)-like(d73/76)、A/chicken/Shanghai/F/1998(H9N2)-like(F/98)和A/chicken/Korea/38349-p96323/1996(H9N2)-like(Kor/ 323/96)及其相似序列做为参比序列构建系统进化树[2],参比序列从NCBI和GISAID中下载。
6.基因序列:全基因组序列使用BioEdit 7.0.9软件修齐,并用MEGA 7.0软件及Neighbor-Joining邻接(NJ)法构建系统发生进化树,采用Bootstrap法(replication=1 000)进行检验。
7.氨基酸序列:通过序列格式转换程序将基因序列转换为FASTA格式,使用BioEdit 7.0.9软件寻找ORF,使用Translate程序,将序列翻译成蛋白质序列,并用MEGA 7.0软件对齐氨基酸序列后,导入BioEdit软件进行氨基酸位点比对分析。
结果1.流行病学调查:病例男性,4岁;2016年8月24日出现发热(38.8 ℃)、流鼻涕、咳嗽、咽痛、乏力等症状,8月25日前往嘉峪关市第一人民医院门诊就诊,医院按照流感样病例采集患者咽拭子进行病毒核酸检测,并给予金莲花颗粒、头孢克圬干混悬剂治疗,病情好转。8月30日流感病毒H9核酸阳性,8月31日进行流行病学调查,再次采集咽拭子标本,流感病毒核酸转为阴性,患儿痊愈,病程7 d。9月1日省级流感参比中心复核,确认为H9N2禽流感病毒核酸阳性。流行病学调查发现,患儿发病前1周跟随母亲去过活禽市场,未直接接触禽类,所有密切接触者也未出现相关症状。9月5日标本接种于形态良好的MDCK细胞进行分离培养,传代2次之后经HA鉴定滴度为1:8,H3为阴性。PCR鉴定为H9N2禽流感病毒核酸阳性。
2.2014 -2019年甘肃省H9N2禽流感病毒的外环境监测:2014年1月至2019年5月,甘肃省活禽市场共采集15 120份外环境样品,检出H9N2禽流感病毒阳性率为23.88%(3 611/15 120)。2014年以来,外环境的阳性标本构成比均高于92.03%。序列信息均上传至国家流感中心病毒基因序列数据库。见表 1。
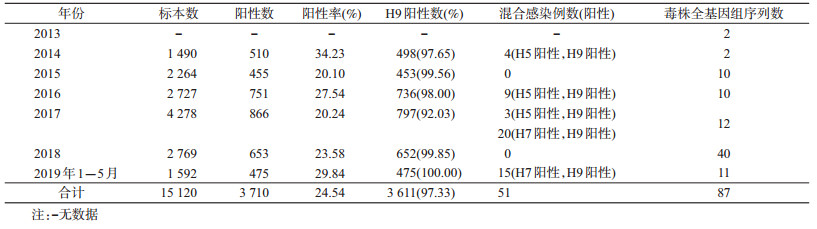
3.同源性分析及各基因片段系统进化分析:将GS_JYG/1397与2013-2019年甘肃省外环境监测中分离获得的87株H9N2禽流感病毒8个基因片段进行相似度比对,发现该毒株HA、NA、MP、NP、NS、PA、PB1和PB2各个基因片段与2013-2019年甘肃省外环境监测H9N2禽流感病毒各基因片段相似度分别为92.5%~97.8%、0.7%~99.1%、94.3%~98.9%、92.4%~98.6%、90.7%~96.3%、93.2%~97.4%、90.8%~94.6%和94.4%~97.6%。分析GS_JYG/1397与2013-2019年甘肃省外环境分离毒株以及1998-2019年公共数据库H9N2禽流感病毒8个基因片段系统进化关系,发现甘肃省人源、禽源和外环境分离的H9N2禽流感病毒的HA基因属于BJ/94-like支系,PB2和MP属于G1/97-like支系,PB1、PA、NS和NP基因属于F/98-like支系,NA基因归属于BJ/94-like支系继续进化的3个分支之一。2018年外环境标本的NP蛋白与高致病性H7N9处于相同的进化分支。GS_JYG/1397的HA基因与甘肃省、四川省、宁夏回族自治区、新疆维吾尔自治区的环境及禽间H9N2相似度较高;GS_JYG/1397的NA基因与甘肃省、四川省和新疆维吾尔自治区的环境及禽间H9N2亲缘关系较近;GS_JYG/1397的MP基因与甘肃省、宁夏回族自治区环境、禽间H9N2、2013年我国台湾地区感染的H9N2和广东省高致病性H7N9禽流感病毒相似度较高;GS_JYG/1397的NP基因与甘肃省环境及湖南省首例人感染H9N2禽流感病毒亲缘关系较近;GS_JYG/1397的NS基因与四川省、宁夏回族自治区的环境及禽间H9N2禽流感病毒相似度较高;GS_JYG/1397的PA基因与四川省环境及禽间H9N2、湖南省首例人感染H9N2禽流感病毒亲缘关系较近;GS_JYG/1397的PB1基因与四川省环境及禽间H9N2禽流感病毒相似度较高;GS_JYG/1397的PB2基因与甘肃省、四川省环境及禽间H9N2禽流感病毒、2016-2017年广东省分离的4株人感染高致病性H7N9禽流感病毒及江西省H10N8病毒亲缘关系较近。见图 1,2。

|
| 注:红色粗体为甘肃省人感染H9N2禽流感病毒;红色为甘肃省外环境H9N2禽流感病毒 图 1 H9N2禽流感病毒HA片段进化树 |

|
| 注:红色粗体为甘肃省人感染H9N2禽流感病毒;红色为甘肃省外环境H9N2禽流感病毒 图 2 H9N2禽流感病毒NA片段进化树 |
4.氨基酸位点分析:甘肃省分离的人源、禽源和外环境的H9N2禽流感病毒蛋白分裂位点为PARSSR↓GLF、PSKSSR↓GLF和PSRSSR↓GLF 3种形式,主要motif形式为PSRSSR↓GLF,均没有发现多个连续的碱性氨基酸插入。2016年外环境分离的H9N2禽流感病毒HA糖基化位点和该病例一致,均为7个。本研究87株外环境H9N2禽流感病毒有11株含有8个糖基化位点,另外76株含有7个糖基化位点,且甘肃省人源和2013-2016年H9N2禽流感病毒在218或220位置的N或T发生突变,导致糖基化位点缺失。外环境和人源的H9N2亚型禽流感病毒受体结合位点结合183位置全部发生H183N突变,226位点全部发生Q226L突变,189位点存在T、D和E 3种氨基酸形式,人源和环境中H9N2 226~228位点为LMG形式,2株禽源的H9N2 226~228位点分别为LMG和LQG形式。
甘肃省从人源、外环境和禽源分离的H9N2禽流感病毒茎部62~64位均缺失I、T、E 3个氨基酸。仅1株病毒发生N142S突变,所有毒株均未发现奥司他韦或扎那米韦耐药位点R292K的突变。
人源和外环境H9N2均发生M2-S31N突变;外环境1株PB2蛋白R318K突变;PB1蛋白的R327K突变在外环境4株H9N2亚型禽流感病毒中均有发现。PA-100A、PA-356R和PA-409N是已适应人类的禽流感病毒分子特征,本研究87株H9N2毒株全部有PA-356R,83株有PA-409N分子特征,但仅6株外环境H9N2禽流感病毒株携带PA-100A突变。同时,发现甘肃省分离的所有H9N2禽流感毒株均携带NS1-42S突变。见表 2。
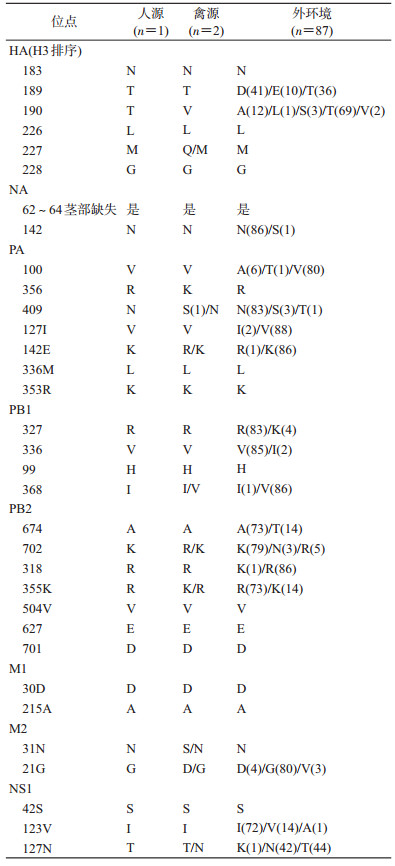
2014年以来,甘肃省外环境H9N2禽流感病毒在外环境阳性标本中构成比均>90%,提示人感染H9N2禽流感病毒发生重配的概率较大,且风险持续存在[6]。该例H9N2禽流感病毒病例的6个内部基因与外环境分离的H9N2内部基因均属于同一个分支,但是PB1与外环境中H9N2属于同一分支的不同进化亚支,而与四川省2013年分离的1株H9N2亲缘关系更近;NS与宁夏回族自治区和四川省禽类中流行的H9N2亲缘性较近。该例人源H9N2和87株外环境H9N2、2013-2019年禽间的H9N2禽流感病毒均为系间多次重配病毒[7],且稳定持续地在禽间传播,污染环境,偶发感染人类[8],需密切关注这种趋势变化。
GS_JYG/1397全基因组进化分析发现,病毒外部基因与当前禽间流行的H9N2禽流感病毒高度相似,且部分内部基因与近年感染人类的H10N8、H7N9禽流感病毒处于共同的分支内,相似度较高,这些毒株均来自于活禽频繁交易的宁夏回族自治区、四川省等地。基因进化分析显示,PB2和M基因源自于G1类似株,中国香港地区的人感染H5N1病毒内部基因与G1系高度同源[9],因此,很可能在西北地区重配其他基因片段,为形成高致病性禽流感病毒提供基本条件。且甘肃省部分外环境标本PA蛋白的V100A、K356R和S409N均具有人流感病毒分子特征[10]。提示,在适宜条件下,禽流感病毒能形成新病原,威胁人类健康[11]。部分环境样本中NS片段发生P42S、T127N突变,提高聚合酶复制能力和对小鼠的致病力[12]。NP蛋白分化了3个分支,其中1个分支和2018年甘肃省外环境和高致病性H7N9禽流感病毒相似度较高,很可能2019年甘肃省人感染H7N9禽流感病毒病例,重配了本地循环的H9N2的NP基因片段[9]。NS和PB1基因片段与宁夏回族自治区和四川省相似度较高,这种高度相似病毒基因片段在西北地区的流行、各省之间活禽交易频繁、部分地区家畜和家禽混合饲养的模式,都会促进禽流感病毒多个基因片段的进化。2019年4月甘肃省发现的1例高致病性人感染H7N9禽流感病毒病例有可能与此有关。
人和外环境的H9N2禽流感病毒均发生M2蛋白S31N突变,表明这些毒株均对于金刚烷胺类耐药[13]。GS_JYG/1397 HA蛋白发生Q226L、H183N、E190T突变,表明与呼吸道飞沫传播密切相关[12]。NS1-42S、PB1-368I、PB2-355K、PB2-504V和M1-30D等突变均具有病毒毒力增强特征,导致禽流感病毒的致病力增加[14],更易感染人类,如果病毒基因突变占据优势的话,将会形成潜在的流感大流行的风险[14],具有重要的公共卫生学意义。
本研究存在不足。由于禽间数据的不足无法更加明确地追踪感染源,在one health理念大力被倡导的今天[15],需要多部门加强合作交流和数据共享,为人类流感大流行做充足准备。
近年来,甘肃省环境中H9N2禽流感病毒监测阳性率居高不下,且该病毒抗原性和宿主特异性不断变化,对其基因特征研究非常重要。
综上所述,该例人感染H9N2禽流感病毒病例为偶发感染,但甘肃省外环境中分离的H9N2禽流感病毒具有一系列哺乳动物适应性分子标记,提示人群感染风险较高。需进一步提高流感监测网络的特异性和敏感性,尽早发现新亚型病毒,追踪感染来源。
利益冲突 所有作者均声明不存在利益冲突
志谢 中国CDC病毒病预防控制所国家流感中心工作人员的大力支持以及嘉峪关市同仁的密切配合
| [1] |
Lu JH, Liu XF, Shao WX, et al. Phylogenetic analysis of eight genes of H9N2 subtype influenza virus:a mainland China strain possessing early isolates' genes that have been circulating[J]. Virus Genes, 2005, 31(2): 163-169. DOI:10.1007/s11262-005-1790-1 |
| [2] |
Gu M, Xu LJ, Wang XQ, et al. Current situation of H9N2 subtype avian influenza in China[J]. Vet Res, 2017, 48(1): 49. DOI:10.1186/s13567-017-0453-2 |
| [3] |
Pu J, Wang SG, Yin YB, et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus[J]. Proc Natl Acad Sci USA, 2015, 112(2): 548-553. DOI:10.1073/pnas.1422456112 |
| [4] |
Fusaro A, Monne I, Salviato A, et al. Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications[J]. J Virol, 2011, 85(16): 8413-8421. DOI:10.1128/JVI.00219-11 |
| [5] |
中国国家流感中心.全国流感监测技术指南(2017年版)[EB/OL]. (2017-09-30)[2019-06-01]. http://www.chinaivdc.cn/cnic/fascc/201802/t20180202_158592.htm. Chinese National Influenza Center. National Influenza Surveillance Technical Guide (2017 Edition)[EB/OL]. (2017-09-30)[2019-06-01]. http://www.chinaivdc.cn/cnic/fascc/201802/t20180202_158592.htm. |
| [6] |
Xu KM, Smith GJD, Bahl J, et al. The genesis and evolution of H9N2 influenza viruses in poultry from southern China, 2000 to 2005[J]. J Virol, 2007, 81(19): 10389-10401. DOI:10.1128/JVI.00979-07 |
| [7] |
何军, 刘丽萍, 侯赛, 等. 安徽省2株人感染H9N2流感病毒基因特征[J]. 中华流行病学杂志, 2016, 37(5): 708-713. He J, Liu LP, Hou S, et al. Genetic characteristics of two humans infected with H9N2 influenza virus in Anhui Province[J]. Chin J Epidemiol, 2016, 37(5): 708-713. DOI:10.3760/cma.j.issn.0254-6450.2016.05.025 |
| [8] |
Zhu R, Xu DW, Yang XQ, et al. Genetic and biological characterization of H9N2 avian influenza viruses isolated in China from 2011 to 2014[J]. PLoS One, 2018, 13(7): e0199260. DOI:10.1371/journal.pone.0199260 |
| [9] |
Yu DS, Xiang GF, Zhu WF, et al. The re-emergence of highly pathogenic avian influenza H7N9 viruses in humans in mainland China, 2019[J]. Euro Surveill, 2019, 24(21). DOI:10.2807/1560-7917.ES.2019.24.21.1900273 |
| [10] |
Han L, He WJ, Yan HX, et al. The evolution and molecular characteristics of H9N2 avian influenza viruses in Jiangxi of China[J]. J Med Virol, 2019, 91(4): 711-716. DOI:10.1002/jmv.25363 |
| [11] |
Jiao PR, Tian GB, Li YB, et al. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice[J]. J Virol, 2008, 82(3): 1146-1154. DOI:10.1128/JVI.01698-07 |
| [12] |
Chang TT, Sun MF, Chen HY, et al. Key features for designing M2 proton channel anti swine flu inhibitors[J]. J Taiwan Inst Chem Eng, 2011, 42(5): 701-708. DOI:10.1016/j.jtice.2011.01.006 |
| [13] |
Jefferson T, Rivetti A, Di Pietrantonj C, et al. Vaccines for preventing influenza in healthy children[J]. Evid-Based Child Health, 2006, 1(2): 367-522. DOI:10.1002/ebch.24 |
| [14] |
Peiris JSM, Guan Y, Markwell D, et al. Cocirculation of avian H9N2 and contemporary "Human" H3N2 influenza A viruses in pigs in southeastern China:potential for genetic reassortment?[J]. J Virol, 2001, 75(20): 9679-9686. DOI:10.1128/JVI.75.20.9679-9686.2001 |
| [15] |
Wan HQ, Sorrell EM, Song HC, et al. Replication and transmission of H9N2 influenza viruses in ferrets:Evaluation of pandemic potential[J]. PLoS One, 2008, 3(8): e2923. DOI:10.1371/journal.pone.0002923 |