 2020, Vol. 41
2020, Vol. 41文章信息
- 曹蓝, 刘艳慧, 李魁彪, 陆剑云, 鲁恩洁, 陈艺韵, 夏丹, 狄飚, 张周斌.
- Cao Lan, Liu Yanhui, Li Kuibiao, Lu Jianyun, Lu Enjie, Chen Yiyun, Xia Dan, Di Biao, Zhang Zhoubin
- 广州市2014-2019年H5亚型禽流感病毒流行与基因遗传特征分析
- Epidemiological and genetic characteristics of H5 subtype avian influenza virus in Guangzhou, 2014-2019
- 中华流行病学杂志, 2020, 41(7): 1115-1120
- Chinese Journal of Epidemiology, 2020, 41(7): 1115-1120
- http://dx.doi.org/10.3760/cma.j.cn112338-20190730-00565
-
文章历史
收稿日期: 2019-07-30




2. 广州市疾病预防控制中心传染病预防控制部 510440;
3. 广州市疾病预防控制中心 510440
2. Department of Infectious Disease Prevention and Control, Guangzhou Municipal Center for Disease Control and Prevention, Guangzhou 510440, China;
3. Guangzhou Municipal Center for Disease Control and Prevention, Guangzhou 510440, China
近年H5亚型禽流感给我国养殖业和公共卫生带来严重威胁,根据HA(hemagglutinin,血凝素)基因进化特点,H5亚型禽流感病毒可以分为Clade 0~9不同进化分支。2014年Clade 2.3.4.4高致病性H5Nx禽流感病毒在世界范围内野鸟和家禽中流行,2014-2015年该分支病毒进化为新型重组病毒引起了人的严重感染[1]。研究显示,新型H5Nx都是以Clade 2.3.4.4为骨架与低致病性禽流感重组而来[2-3]。该分支进一步可分为A~D 4个组,其中Clade 2.3.4.4.C H5N6病毒宿主来源多样,包括野鸟、家禽、鸽子、猪、猫和人[4]。2013年开始,Clade 2.3.4.4.C H5N6禽流感在我国流行[5-6],2016年秋,在韩国和日本流行[7]。Yu等[4]对2014-2016年我国活禽市场监测的数据显示,H5N6禽流感已经取代了H5N1成为在华南地区主要流行的亚型。2014年12月广东省确诊首例人感染H5N6禽流感病例[8],有研究显示,不同病例H5N6病毒内部基因的进化来源不同[9],提示H5N6禽流感病毒遗传背景复杂[10],因此开展新型H5N6禽流感病毒分子流行病学监测尤为重要。
材料与方法1.标本来源:
(1)采样时间与地点:参照监测方案《广州市人禽流感监测方案(2013版)》《广州市人禽流感监测方案(2016版)》和《广州市禽类市场环境禽流感病毒污染监测方案》,2014年1月至2017年7月在广州市11个区各选定2个禽类市场,每季度在每个市场采集10份外环境涂抹标本;2017年8月至2019年3月在11个区的所有社区卫生服务中心管辖范围内均选定1个禽类市场,每个市场选择1个禽类经营档口作为采样点,每个档口采集5份标本,当年11月至次年5月每月完成两次监测,当年6-10月每月完成一次监测。
(2)采样方法:活禽非限制区采集标本类型主要包括禽类粪便、笼具涂抹、禽类饮水、污水、脱毛机内壁、砧板与刀具、污物桶等;活禽限制区采集标本类型主要包括冰柜内壁、禽肉涂抹、销售展台、砧板与刀具、污物桶、售卖区污水及储存光禽的容器(活禽宰杀去毛后未经过冷链系统运输或储存的称为光禽)等。人感染禽流感病例咽拭子标本由广州市各级医疗机构对接诊的有禽类接触史的发热并怀疑感染禽流感的患者,采集咽拭子。上述各类型标本4 ℃保存,在24 h内送至实验室于-80 ℃保存备检。
2.标本检测:标本冻融后置于振荡器充分震荡,提取病毒RNA核酸。采用实时荧光定量RT-PCR方法进行通用A型禽流感病毒核酸检测,阳性标本进行H5亚型病毒核酸检测。病毒核酸提取试剂盒和禽流感病毒检测试剂盒(荧光定量PCR法)分别购于德国Qiagen公司和中国江苏硕世生物科技股份有限公司。
3.基因序列扩增与测序:应用Oligo 6软件设计H5N6禽流感病毒HA基因和神经氨酸酶(NA)基因全长扩增引物,通过简单随机抽样方法选取2014-2019年46份环境H5阳性标本和2份H5N6阳性病例咽拭子标本,通过RT-PCR进行基因扩增,经毛细管电泳初步鉴定后,将阳性扩增产物送至广州市华大基因公司进行测序。
4. HA和NA基因分子特征分析:应用DNA Star 7.1软件拼接HA和NA基因序列,并对序列进行核苷酸同源性分析。用MegAlign软件对重要蛋白位点(受体结合位点、抗原位点、裂解位点、糖基化位点、耐药位点等)进行氨基酸插入、缺失和突变分析。
5.基因遗传进化分析:从美国国立生物技术信息中心下载国内外地区人源、禽源和环境H5和N6亚型基因序列作为参比序列。采用Mega 6.0软件,以基因开放阅读框为基本单元,绘制HA和NA基因遗传进化树,分析各基因的分子流行进化特点。绘制方法为Neighbor-joining法(参数设置为1 000 replications)及Maximum composite likelihood model比对核苷酸序列。
结果1.总体情况:2014年1月至2019年3月累计检测禽类市场外环境标本52 284份,检出H5阳性标本1 094份,阳性率为2.09%,2018年最高(2.87%),2014年最低(0.70%),各年H5阳性构成比在3.71%~12.14%之间。见表 1。2014-2016年广州市禽类市场禽流感病毒流行以季节性升高为主要特点,冬春季节出现流行高峰。2017年8月实施环境禽流感全覆盖监测后,流行趋势趋于稳定。总体上,H5亚型禽流感病毒持续流行,平均阳性率为2.09%,2017年7月检出率最高(21.11%)。广州市第1例和第2例H5N6确诊病例均出现在冬春季节流行高峰期,第3例出现在全覆盖监测实施后。

2. HA和NA基因遗传进化分析:2014-2019年广州市环境H5亚型禽流感均属于Clade 2.3.4.4.C分支,以时间聚类为特点,2014-2016年毒株归属于同一进化分支,2017-2019年毒株归属于另一进化分支,见图 1。N6基因主要归属于欧亚谱系H6N6进化分支,该分支内年代相近的毒株聚为一类,形成3个独立小分支。发现1株病毒NA基因与H4N6禽流感病毒亲缘关系较近,归属于同一进化分支。见图 2。

|
| 图 1 2014-2019年广州市禽流感病毒H5基因遗传进化树 |

|
| 图 2 2014-2019年广州市禽流感病毒N6基因遗传进化树 |
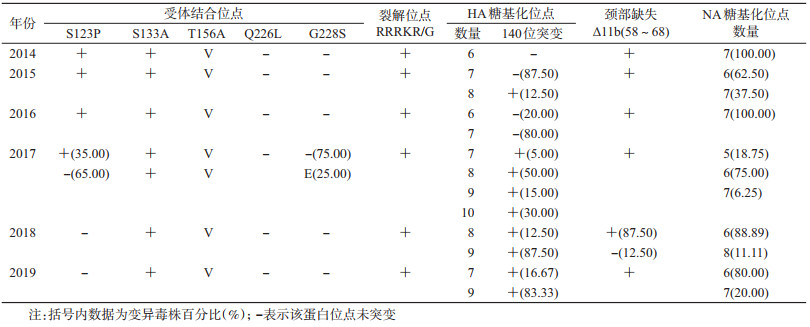
3. HA和NA基因分子特征分析:所有毒株在受体结合位点226位未发生突变,有5株病毒在228位发生G228E突变。HA蛋白糖基化位点主要位于26、27、39、140、181、302、499和558位,2017-2019年毒株均出现由氨基酸缺失导致的糖基化位点增加(140-NHT)。HA蛋白抗原位点变异主要出现在B、E区,并且以时间分布为特点:B区129位,2014-2015年毒株为L,2016-2019年毒株为S;B区183位,2014-2016年毒株为N,2017-2019年毒株为S;B区189位,2014-2017年毒株为N,2018-2019年毒株为D;E区269位,2014-2016年毒株M,2017-2019年毒株为I。A区抗原位点变异相对较少,但表现较为明显的多样性,C区和D区抗原位点相对保守。NA蛋白变异分析结果显示,除H4N6分支病毒外,所有毒株均出现颈部11个氨基酸缺失,糖基化位点主要位于51、70、146、201和402位,所有毒株均未出现H274Y和R292K耐药变异。见表 2。
目前广州市已将禽类市场外环境禽流感病毒流行纳入长期监测工作,总体上通用A型禽流感病毒阳性率为19.89%,低于福建省的监测数据[11],而2006-2012年广州市职业暴露人群血清抗体监测显示,H5抗体平均阳性率为0.22%,提示职业人群存在感染禽流感病毒的风险[12]。
2014-2019年H5亚型禽流感病毒长期流行,阳性率在0.70%~2.87%之间(按年份),平均阳性率为2.09%,低于福建省的监测数据[11]。但各年H5阳性构成比逐年增加,提示H5亚型在环境各亚型禽流感病毒中所占比例有上升趋势。2018年9月广州市确诊了第3例人感染H5N6禽流感病例,前期研究显示,广州市H5N6禽流感病例具有禽类接触史[8],本研究结果也显示,人源毒株与市场外环境H5N6病毒同源性较高,属于同一进化分支,进一步证实禽类市场是感染来源[13]。
基因进化分析显示,2014-2019年广州市H5N6流行毒株与国内毒株亲缘关系较近,位于Clade 2.3.4.4.C分支,属于近年广东省主要流行分支[14]。进一步分析发现,不同年份流行毒株HA和NA基因均呈现以时间为聚类的进化特点,形成不同的进化分支。同时抗原位点变异分析也发现,与云南省研究结果不同[15],广州市H5N6抗原位点变异主要发生在B、E区,且抗原位点变异呈现较为明显的时间一致性,同一年份流行株常发生与上一年份流行株不同的抗原位点变异,提示广州市H5N6病毒在持续进化和变异。2株人源病毒NA基因位于不同的进化分支内,推测进化起源可能不同。另有1株2018年病毒位于H4N6进化分支,提示N6基因进化的多样性,同时也提示可能存在H5N6病毒与H4N6重组的风险。有研究表明,H5N6禽流感病毒可与低致病性禽流感病毒发生重组[16],因此应加强H5N6与低致病性禽流感病毒的重组监测。
受体结合位点分析显示,广州市流行株仍为禽源受体,但普遍发生的S123P、S133A和T156A位点突变可以提高病毒与人源受体的结合能力。研究还发现,2014-2019年毒株普遍存在糖基化位点数量上和位置上的变异,总体上HA蛋白糖基化位点为6~10个,NA蛋白糖基化位点为5~8个,其中以2017年毒株变异最大。从2017年起,所有毒株发生142位氨基酸缺失导致的糖基化位点的增加(140-NHT),Chen等[17]在对H5N6新型重组病毒研究时也发现,与H9N2/H7N9病毒重组的H5N6病毒均携带有这种由氨基酸缺失所致的140位糖基化位点增加,这种变异是否会影响病毒传播和致病能力,有待进一步研究。
本研究对广州市2014-2019年禽类市场外环境H5亚型禽流感病毒的流行和基因遗传变异情况进行监测和分析,发现H5亚型禽流感病毒长期流行,HA和NA基因呈现以时间为聚类的进化特点,糖基化位点和抗原位点的变异也呈现时间分布的特点,且流行株普遍出现与人源受体结合能力增强的突变,提示病毒在持续进化和变异。本研究仅随机选取48份标本进行HA和NA基因的测序分析,对于全基因组变异和进化的系统分析仍有待进一步研究。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
Yang L, Zhu WF, Li XD, et al. Genesis and dissemination of highly pathogenic H5N6 avian influenza viruses[J]. J Virol, 2017, 91(5): e02199-16. DOI:10.1128/JVI.02199-16 |
| [2] |
Bi YH, Mei K, Shi WF, et al. Two novel reassortants of avian influenza A(H5N6) virus in China[J]. J Gen Virol, 2015, 96(5): 975-981. DOI:10.1099/vir.0.000056 |
| [3] |
Xu HY, Meng F, Huang DH, et al. Genomic and phylogenetic characterization of novel, recombinant H5N2 avian influenza virus strains isolated from vaccinated chickens with clinical symptoms in China[J]. Viruses, 2015, 7(3): 887-898. DOI:10.3390/v7030887 |
| [4] |
Yu BH, Quan CJ, Wang QL, et al. Genesis, Evolution and prevalence of H5N6 avian influenza viruses in China[J]. Cell Host Microbe, 2016, 20(6): 810-821. DOI:10.1016/j.chom.2016.10.022 |
| [5] |
Du YY, Chen MY, Yang JY, et al. Molecular evolution and emergence of H5N6 avian influenza virus in central China[J]. J Virol, 2017, 91(12): e00143-17. DOI:10.1128/JVI.00143-17 |
| [6] |
Qi X, Cui LB, Yu HY, et al. Whole-genome sequence of a reassortant H5N6 avian influenza virus isolated from a live poultry market in China, 2013[J]. Genome Announc, 2014, 2(5): e00706-14. DOI:10.1128/genomeA.00706-14 |
| [7] |
Okamatsu M, Ozawa M, Soda K, et al. Characterization of highly pathogenic avian influenza virus A(H5N6), Japan, November 2016[J]. Emerg Infect Dis, 2017, 23(4): 691-695. DOI:10.3201/eid2304.161957 |
| [8] |
Li KB, Liu H, Yang ZC, et al. Clinical and epidemiological characteristics of a patient infected with H5N6 avian influenza A virus[J]. J Clin Virol, 2016, 82: 20-26. DOI:10.1016/j.jcv.2016.06.004 |
| [9] |
Xu W, Li XD, Bai T, et al. A fatal case of infection with a further reassortant, highly pathogenic avian influenza (HPAI) H5N6 virus in Yunnan, China[J]. Infect Genet Evol, 2016, 40: 63-66. DOI:10.1016/j.meegid.2016.02.020 |
| [10] |
Lee DH, Bertran K, Kwon JH, et al. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx Clade 2.3.4.4[J]. J Vet Sci, 2017, 18 Suppl 1: 269-280. DOI:10.4142/jvs.2017.18.S1.269 |
| [11] |
陈平, 谢剑锋, 林琦, 等. 福建省人感染H5N6禽流感病毒与外环境禽流感病毒相关性分析[J]. 中国人兽共患病学报, 2018, 34(8): 721-727, 732. Chen P, Xie JF, Lin Q, et al. Molecular and phylogenitic analysis of avian influenza A (H5N6) between strain isolated from human case and avian influenza viruses of 5 years environmental surveillance, Fujian, China[J]. Chin J Zoon, 2018, 34(8): 721-727, 732. DOI:10.3969/j.issn.1002-2694.2018.00.136 |
| [12] |
陈宗遒, 陆剑云, 肖新才, 等. 广州地区2006-2012年人感染H5/H7/H9亚型禽流感病毒风险监测[J]. 中华流行病学杂志, 2013, 34(9): 900-905. Chen ZQ, Lu JY, Xiao XC, et al. Evaluation on the risks of H5, H7 and H9 avian influenza infections in Guangzhou:using data from the 2006-2012 avian influenza surveillance program[J]. Chin J Epidemiol, 2013, 34(9): 900-905. DOI:10.3760/cma.j.issn.0254-6450.2013.09.010 |
| [13] |
Zhang RS, Chen TM, Ou XH, et al. Clinical, epidemiological and virological characteristics of the first detected human case of avian influenza A(H5N6) virus[J]. Infect Genet Evol, 2016, 40: 236-242. DOI:10.1016/j.meegid.2016.03.010 |
| [14] |
Yuan RY, Wang Z, Kang YF, et al. Continuing reassortant of H5N6 subtype highly pathogenic avian influenza virus in Guangdong[J]. Front Microbiol, 2016, 7: 520. DOI:10.3389/fmicb.2016.00520 |
| [15] |
孔强, 赵焕云, 田康, 等. 2014-2015年云南省边境H5N6禽流感病毒血凝素与神经氨酸酶基因序列分析[J]. 中华预防医学杂志, 2016, 50(12): 1105-1107. Kong Q, Zhao HY, Tian K, et al. Molecular analysis of hemagglutinin and neuraminidase genes of avian influenza virus H5N6 subtype in the border of Yunnan Province from 2014 to 2015[J]. Chin J Prev Med, 2016, 50(12): 1105-1107. DOI:10.3760/cma.j.issn.0253-9624.2016.12.016 |
| [16] |
Zhang Y, Chen MM, Huang YW, et al. Human infections with novel reassortant H5N6 avian influenza viruses in China[J]. Emerg Microbes Infect, 2017, 6(6): e50. DOI:10.1038/emi.2017.38 |
| [17] |
Chen LJ, Lin XD, Tian JH, et al. Diversity, evolution and population dynamics of avian influenza viruses circulating in the live poultry markets in China[J]. Virology, 2017, 505: 33-41. DOI:10.1016/j.virol.2017.02.009 |