 2020, Vol. 41
2020, Vol. 41文章信息
- 龙智平, 王帆.
- Long Zhiping, Wang Fan
- 多组学整合分析的设计及统计方法在肿瘤流行病学研究中的应用
- Study design and statistical methods used for integrative analysis on multi-omics in cancer epidemiology
- 中华流行病学杂志, 2020, 41(5): 788-793
- Chinese Journal of Epidemiology, 2020, 41(5): 788-793
- http://dx.doi.org/10.3760/cma.j.cn112338-20190624-00461
-
文章历史
收稿日期: 2019-06-24




“组学”技术广泛应用于肿瘤的研究,从分子学角度看,组学是对一组分子进行全面或全局的评估[1]。肿瘤是一类分子机制复杂的疾病,高通量的多组学数据可以很好地解读肿瘤分子图谱,但单一类型的组学研究只能在单一水平上揭示肿瘤相关分子信息,例如:基因组学只能揭示癌症患者基因改变的情况,但并不是所有的基因变异都会导致表达及功能的改变。要了解肿瘤起始、进展和转移扩散的机制,开发新的肿瘤生物标志物,以及发现新的治疗干预的靶点,我们需要在多个层面采用多种组学策略。多组学整合分析是指对来自不同组学如基因组学、转录组学、蛋白质组学和代谢组学的数据进行归一化处理、比较分析,建立不同组间数据的关系,综合多组学数据对生物过程从基因、转录、蛋白和代谢水平进行深入的阐释,从而全面了解生物系统[2]。与单组学研究相比,多组学分析有可能更深入地了解肿瘤从一个组学级别到下一个组学级别信息流的变化[3]。
随着高通量测序、质谱法以及计算机科学和算法的发展,组学数据研究的障碍逐渐消除,对肿瘤的探索有更广阔的空间。人类基因组受到多个组学层面的调节[4],如何整合各个组学的信息从而形成系统层面的理解将是一个挑战。本文将讨论目前肿瘤学背景下的组学数据生成及应用,在此基础上,着重总结多组学研究的设计以及数据分析和整合的策略,为肿瘤流行病学的发展提供依据。
1.组学数据的生成:在过去的几十年中,组学技术提供了大量从基因到代谢方面的肿瘤特异性信息,为肿瘤的预防和治疗特别是个性化医疗提供了依据(表 1)。
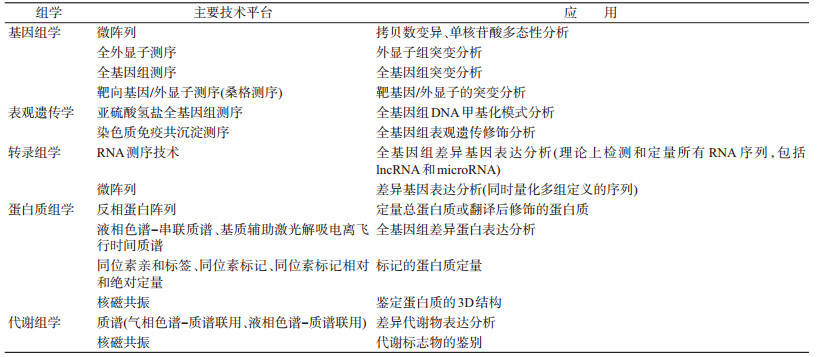
(1)基于二代测序(NGS)技术的基因组学、表观遗传学和转录组学:癌症本质上是基因组的疾病[5],在NGS技术的快速发展下[6],探索癌症基因组改变及其多样性、全面分析癌症基因组谱已成为可能。到目前为止,全球已有超过5万个癌症基因组被测序[7-8]。大规模基因组研究和NGS技术揭示了关于肿瘤异质性的更多生物学细节,对特定分子生物标志物的选择和临床决策具有重要意义。
表观遗传学是在基因组的水平上,研究表观遗传化学修饰,例如DNA的甲基化和乙酰化[9]。染色质免疫共沉淀测序(ChIP-seq)技术被广泛用于表观遗传学分析,ChIP-seq技术由染色质免疫共沉淀技术与NGS相结合产生[10],其应用已深入到各种癌症发生和进展的基因调控事件中[11]。此外,全基因组亚硫酸氢盐测序亦基于NGS技术而发展[12]。通过上述技术产生的多个数据集被收集在DNA甲基化数据库MethyCancer和组蛋白修饰数据库HIstome中,表观遗传学研究在揭示与肿瘤相关的表观遗传标记中发挥了重要作用。
转录组学是通过使用高通量方法产生关于基因组或特定细胞在某一发育阶段或功能状态下的总转录物的信息,这些转录物包括mRNA和非编码RNA(如microRNA、lncRNA、circRNAs等)。常用的转录组学技术有微阵列和RNA测序技术(RNA-Seq)。微阵列是NGS技术出现之前的常规实验技术[13],RNA-Seq是基于NGS技术的高通量大规模平行测序方法[14],近年来RNA-Seq已成为最强大和全面的转录组分析技术,几乎取代了所有表达芯片。转录组分析的应用涉及广泛的肿瘤研究,包括肿瘤诊断、分类、监测以及对治疗的反应[15]。
(2)基于质谱技术的蛋白质组学和代谢组学:蛋白质组学是鉴定和定量生物样品中某一时间存在的蛋白质的研究,包括蛋白质修饰、蛋白质之间的相互作用、蛋白质结构描述等。
在过去的40年中,基于抗体的反向蛋白阵列(RPPA)技术被广泛用于细胞系和肿瘤组织的蛋白质组学研究[16-17]。癌症蛋白质组图谱(TCPA)使用RPPA技术对癌症和肿瘤基因图谱(TCGA)中大量肿瘤样品和细胞系进行检测,所鉴定的蛋白质几乎涵盖了所有肿瘤相关的主要通路[18]。但由于抗体的非特异性和低通量[18],不足以应对复杂蛋白质混合物的完全检测,因此具有高通量、高特异性、高灵敏度特征的质谱技术成为了蛋白质组学研究的广泛而通用的技术。
近年来,高分辨率Orbitrap质谱仪器与MaxQuant等计算平台相结合,促进了人类细胞和组织中几乎所有表达蛋白(约18 000种蛋白质)的全基因组鉴定和定量[19]。Vasaikar等[20]在临床蛋白质组学肿瘤分析联盟的支持下,发布了一个新的数据门户网站,它将TCGA临床基因组学数据与MS衍生的蛋白质组学数据联系起来,其方式类似于TCPA利用RPPA阵列进行的研究。
代谢物水平是基因活性的另一个指标,也反映了外部环境对生物体的影响。代谢物是细胞代谢活动的产物,例如氨基酸、碳水化合物和脂质,其水平和比例在肿瘤发展中会发生改变,从而反映肿瘤中的异常代谢功能[21]。检测代谢物的两种主要分析方法是核磁共振(NMR)和质谱。虽然NMR被认为是化合物鉴定的金标准并且能够定量,但基于质谱的方法更敏感,能够检测低丰度代谢物,且检测的代谢物更多[22]。
2.多组学研究设计:当在单个研究中应用多个组学技术时,研究设计的选择很重要,因为它将在整个研究中产生许多影响,特别是在数据分析方面。下面将介绍最常见的4种研究设计。
(1)重复性研究设计:重复性研究通常是最便捷的方法,第一个组学研究检测一组样品,然后重复研究方案以产生第二个组学研究的样品(或在多个实验室中平行应用相同的方案以产生多个样本,也可以简单地理解为在不同的时间点或地点重复性的收集样本)。这里的重复性主要是指研究以相同的方法采集或者生成样本,并不是指整合研究设计的重复。例如,Qiu等[23]以相同的方案,从位于中国和美国的4家医院收集227例结直肠癌患者的癌组织和距边缘5 cm的邻近非肿瘤组织样本,其中34例用于转录组学分析,193例用于代谢组学分析。代谢组学分析确定了一组能够预测结直肠癌患者手术和化疗后复发率和存活率的15种显著改变的代谢物,整合转录组学分析表明,这些代谢物的差异表达与癌细胞增长有关。
重复性研究设计的优点是,每一次检测在统计上是独立的,或者每一个组学研究都是对同一假设的独立检验[24]。然而,如Tillinghast[25]所述,这种设计会将批量效应引入数据分析。当在单组学技术的大型研究中出现批量效应时,可以通过统计方法进行校正,例如在组合或者用其他更复杂的方法分析之前,可以对每一批次的数据进行简单的中心化处理[26]。但是,当多个组学技术应用于独立的组学实验研究时,不能进行批次校正。
(2)源匹配研究设计:选择使用生物系统的不同部分进行不同的分析,称为源匹配研究,在多组学研究中,即为对同一研究对象的不同生物样本做不同的组学分析。这种研究设计在癌症研究中很常见,比如,采集肿瘤患者的血液或尿液用于代谢组学分析,同时采集肿瘤组织进行表达谱分析。源匹配研究也可以用于体外细胞实验,例如,细胞材料用于转录组学分析,细胞培养基用于代谢组学。在Robles和Harris[27]的研究中,通过结合基因组学(DNA甲基化)、转录组学(miRNA和mRNA表达)和代谢组学(促炎细胞因子和尿液代谢物)的数据,发现了可以对肺癌进行早期检测和预后评估的生物标志物。
(3)复制匹配的研究设计:通常选择研究设计主要考虑的决定因素是实际操作的可行性。例如,是否可以从单个生物样本或来源获取多个组学的分析样品。在细胞实验中,如果要用代谢组学分析细胞内提取物并通过转录组学分析分离的RNA,那么代谢组学分析需要另外单独的样本,因为RNA提取过程和用于代谢组学的细胞沉淀的样品制备方案是相互排斥的。在这种情况下,可以使用复制匹配的研究设计,即收集相同来源的两个或更多个重复样品并同时制备用于组学分析。当进行复制匹配的研究时,需要注意的是在每个组学研究中样本的随机化,以免在组学研究结果之间引入任何偏差。复制匹配的研究不像重复性研究一样存在批次效应,因为各个组学的样品是同时产生或获得的,避免了批次效应的引入。
(4)分割样本研究设计:除了重复性研究外,其他的研究设计都假设在不同的组学数据集之间产生的数据没有批次效应,然而,批次效应可能仍然存在,特别是如果在一段时间内检测大量的样本。从数据整合角度来看,理想的研究设计是样品来自相同的生物源材料。例如,一块组织可以切成两个部分,一个用于代谢组学分析,而另一个用于提取RNA。这种研究设计称为分割样本研究设计。
在分割样本设计中,数据来自相同的组织并且是同时获取的,这使得我们能够在样本收集的特定时间点探索该特定组织内的疾病机制。在可以自由选择研究设计的情况下,分割样本研究设计将是最佳的,其次是复制匹配研究,源匹配研究可以作为一个备选的设计,源匹配设计说明了不同的组学数据所来自的不同样本源的情况,这种多层次的组学数据可以建立跨组织类型的系统网络,也可以证实研究结果或提高其可信度。但源匹配设计必须慎重考虑如何解释多个数据集之间的生物学联系。如果可能的话,最好避免重复研究设计以规避固有的批次效应。在多组学数据整合的数据集中,TCGA是应用最广泛的一个,患者的癌组织和癌旁组织在收集后,同一组织可以分割后用于不同的组学平台检测[28]。基于TGCA的多组学数据整合的典型例子是由癌症基因组图谱网络公布的关于乳腺癌分子图谱研究[29]。该研究通过6个平台分析了不同组的乳腺肿瘤,从而能够识别亚型特异性改变。通过多组学交叉整合分析,发现4种主要的乳腺癌亚型(管腔A、腔B、基底样和HER2+)具有特定的基因组、蛋白质组和临床特征。
3.多组学数据整合方法:近年来发表了多项使用多组学数据集的癌症相关研究,多组学癌症数据集经常使用统计学独立分析,然后以图形或定性方式进行比较,例如富集分析。Ebbels和Cavill[30]的研究认为数据整合有3个层次:概念整合、统计整合和基于模型的整合。概念整合是用于组学数据整合最早和最简单的方法,而基于模型的整合有待进一步研究,目前被广泛应用的组学数据整合方法主要是统计整合。因此本文主要介绍统计整合相关概念。
(1)概念整合和基于模型的整合:概念整合是指先分别分析多个组学数据集,然后,在没有对整个数据集进行任何进一步分析的情况下对得到的结论进行人工的比较匹配,寻找关联性。如在Fan等[31]的研究中,通过代谢组学和转录组学分析抗癌剂亚硒酸盐对人肺癌A549细胞的作用机制。研究揭示了亚硒酸盐改变的代谢网络,而这种变化又用于推断从转录组学分析获得的上游调节通路的变化。概念整合的方法可以产生有价值的信息,但是,它也会错过在只有同时分析两个数据集时才能找到的关联。
基于模型的整合在目前仍处于起步阶段,难以获得理想的结果。基于模型的整合需要获取全面的组学数据以及对复杂生物系统的高度理解,虽然许多系统部分(例如基因、代谢物)可能是已知的,但它们相互作用以形成完全功能系统的方式通常是未知的。尽管如此,随着组学技术的发展该领域也将成为一个研究的焦点,并且已经出现了许多可使用的技术和建模方法[30],比如多层贝叶斯模型、遗传调控模块、基于约束的重建和分析等。
(2)统计整合:统计整合是最常见的整合形式,这种方法是在不同数据集的元素之间寻求统计关联。统计整合主要有基于相关性的整合、基于多元分析的整合以及基于通路的整合。
基于相关性的整合是寻找一个数据集的元素与另一个数据集的元素之间的相关性。相关性经常被用于检查两个组学数据之间的关联,有许多方法可以评估两组数据的相关性,其中最常见的是Pearson和Spearman相关,此外其他测量相关性的方法有Goodman和Kruskal伽玛检验、稳健的线性模型以及部分相关性等。目前,相关性的直接应用存在许多潜在问题,在通路中密切相关的那些基因、蛋白质和代谢物通常不显示相关性,而相关性也可能在网络上的远距离处发生;仍需要做更多的工作来确定部分相关性是否有助于识别最直接的联系;在许多实验设计中,多个组学的变化不会同时发生,时间问题的存在会模糊组学间数据的联系,某个组学变化可能在更早的时间点与另一个组学的变化相关联。当然,时间问题贯穿整个多组学的整合过程,不仅体现在组学数据的相关性上,从实验的设计到结果的解释,都要考虑时间问题的存在。
基于多元分析的整合利用,如采用偏最小二乘法(PLS)和主成分分析(PCA)等多元统计分析方法对数据进行建模,预测并寻找变量或样本之间的关系,分析数据中固有的变化。当使用多元模型进行组学数据整合时,可以使用一个数据集来预测另一个数据集或者找到两个数据集之间的协方差关联。在Nevedomskaya等[32]的研究中,使用O2PLS建模整合基因表达和代谢物数据,全基因组关联研究引导的NCI-60转录组和代谢组数据的整合鉴定了胞外-5′-核苷酸酶(NT5E或CD73)作为癌细胞中代谢表型的主要决定因素。用于组学数据整合多元统计的方法还有如正交偏最小二乘判别分析(OPLS-DA)、PLS-DA和典型相关分析(CCA)等,这些方法可分为无监督和监督两种类型,无监督的方法(PCA、CCA等)是探索性地分析研究对象的生物学概况,将其分配到不同的亚组,例如可以用于肿瘤分子分型研究;监督的方法(PLS、OPLS-DA等)可以理解为回归分析,如利用被标记的研究对象的生物信息,推导出不同表型的模式,以进行不同表型特征下的预测(如肿瘤亚型和预后)。如何选择合适的方法和对模型结果的解释是目前使用多变量建模进行组学数据整合的挑战。因此,需要开发新的工具使这些方法不仅可以使用,而且可以给出合理的解释,并且非专业的人可以轻松应用,同时进一步明确每种方法的局限性和优势及其附带的可视化。
基于通路的整合是一类使用现有生物学知识通过如KEGG和Wikipathways等数据库中预定义的代谢通路整合数据的方法,这种方法不同于研究反应通量的建模工作,它是以自动化方式将检测到的代谢物、基因和蛋白质映射到通路上,寻找在不同条件之间出现统计学上显著改变的通路或者通路特征与感兴趣的表型终点之间的相关性,这也是一种网络功能研究方法。一系列软件工具可用于基于通路的组学数据整合,其中许多还可以处理多种类型的组学数据。商业产品包括Ingenuity Pathway Analysis(IPA)和Omix,强大的开源解决方案包括Vanted以及MetScape,其他可用的在线工具有Prometra和Paintomics等。Zhang等[33]对来自胰腺肿瘤和邻近健康组织的一组活检样本中代谢物和基因数据进行整合分析,研究利用加权共表达网络分析来鉴定在患者群组中表达模式相似的代谢物簇,再通过通路富集分析(IPA软件)将代谢物分配到已知的代谢通路上,从而能够与在同一代谢通路上的转录物进行直接比较。研究从脂质代谢中鉴定出8种代谢物和4种酶作为潜在的治疗靶点。
4.讨论:由于基因、蛋白质和代谢物所构建的网络复杂性质,组学数据之间的关联方向和强度在实验条件之间变化很大,而这些关联又可能被每个数据集中固有的噪声和方差结构所隐藏,因此组学间的相关性并不像期望的那样显著。但这反过来也使得我们观察到不同的组学数据集具有很多互补的信息内容,因此多组学整合分析有可能揭示更多的生物学信息而不是单一组学分析的总和。
随着多组学研究在投入上的增加和技术上的提高,多组学整合分析方法的应用将继续扩大。尽管基于多元分析的整合方法在模型解释和选择上很复杂,并且结果受到很多因素的影响,但借助于多元分析方法,可以实现对多个组学数据集间的重叠信息进行更好的解读,比如重叠信息在数量上的统计相关性和内容上的生物学联系,以及利用重叠信息建立肿瘤表型的强预测模型。因此这种方法具有很大的潜力,非常有必要进一步研究。对于生物学家来说,基于通路的整合方法是最直观的,并为数据的解释提供了充足的相关信息。然而,需要注意的是,这种方法基于现有的通路知识,不能用于发现新的肿瘤生物学关联,因此需要其他技术来探索这些关联。
要在实践中广泛采用多组学整合分析,必须应对各种分析挑战,特别是那些用于数据整合的统计方法,由于每个数据集都有自己的差异和偏差,需要一个强大且可重复的统计框架来正确分析多个不同的数据集。而同时分析3个或更多的数据集需要更复杂的多维方法,如贝叶斯模型、神经网络或降维。除了组学数据整合分析方法的挑战之外,各组学数据的准确性、重复性以及解读性仍有待提高。个体水平上,全基因组数据集存在固有错误率[34];蛋白质组数据和代谢组数据的固有复杂性和所使用的多个平台技术使得数据转化和标准化以及重复性变得困难[35];而高通量的组学技术不断带来新的分子信息,用现有的生物学技术不足以进行功能的验证。
尽管目前组学数据整合已经得到了普遍重视并取得了一定进展,但不同组学数据的整合仍存在多种问题,使得现有的组学数据还没有被充分解读。大部分研究还集中于数据的预处理和可视化层次,有待寻找在生物学意义上整合多组学数据的新方法。但是,多组学数据的整合是目前最有希望的,可用于揭示肿瘤中所有类别分子之间可能发生的相互作用。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease[J]. Genom Biol, 2017, 18: 83. DOI:10.1186/s13059-017-1215-1 |
| [2] |
Karczewski KJ, Snyder MP. Integrative omics for health and disease[J]. Nat Rev Genet, 2018, 19(5): 299-310. DOI:10.1038/nrg.2018.4 |
| [3] |
Senft D, Leiserson MDM, Ruppin E, et al. Precision oncology:the road ahead[J]. Trends Mol Med, 2017, 23(10): 874-898. DOI:10.1016/j.molmed.2017.08.003 |
| [4] |
Lowe WL, Reddy TE. Genomic approaches for understanding the genetics of complex disease[J]. Genome Res, 2015, 25(10): 1432-1441. DOI:10.1101/gr.190603.115 |
| [5] |
Stratton MR, Campbell PJ, Futreal PA. The cancer genome[J]. Nature, 2009, 458(7239): 719-724. DOI:10.1038/nature07943 |
| [6] |
Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing[J]. Nat Rev Genet, 2010, 11(10): 685-696. DOI:10.1038/nrg2841 |
| [7] |
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways[J]. Nature, 2008, 455(7216): 1061-1068. DOI:10.1038/nature07385 |
| [8] |
The International Cancer Genome Consortium. International network of cancer genome projects[J]. Nature, 2010, 464(7291): 993-998. DOI:10.1038/nature08987 |
| [9] |
Piunti A, Shilatifard A. Epigenetic balance of gene expression by polycomb and COMPASS families[J]. Science, 2016, 352(6290): aad9780. DOI:10.1126/science.aad9780 |
| [10] |
Nakato R, Shirahige K. Recent advances in ChIP-seq analysis:from quality management to whole-genome annotation[J]. Brief Bioinform, 2017, 18(2): 279-290. DOI:10.1093/bib/bbw023 |
| [11] |
Raj U, Aier I, Semwal R, et al. Identification of novel dysregulated key genes in Breast cancer through high throughput ChIP-Seq data analysis[J]. Sci Rep, 2017, 7: 3229. DOI:10.1038/s41598-017-03534-x |
| [12] |
Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation[J]. Epigenet Chromat, 2016, 9: 26. DOI:10.1186/s13072-016-0075-3 |
| [13] |
Heller MJ. DNA microarray technology:devices, systems, and applications[J]. Annu Rev Biomed Eng, 2002, 4: 129-153. DOI:10.1146/annurev.bioeng.4.020702.153438 |
| [14] |
McGettigan PA. Transcriptomics in the RNA-seq era[J]. Curr Opin Chem Biol, 2013, 17(1): 4-11. DOI:10.1016/j.cbpa.2012.12.008 |
| [15] |
Li MY, Sun QR, Wang XS. Transcriptional landscape of human cancers[J]. Oncotarget, 2017, 8(21): 34534-34551. DOI:10.18632/oncotarget.15837 |
| [16] |
Sheehan KM, Calvert VS, Kay EW, et al. Use of reverse phase protein microarrays and reference standard development for molecular network analysis of metastatic ovarian carcinoma[J]. Mol Cell Proteom, 2005, 4(4): 346-355. DOI:10.1074/mcp.T500003-MCP200 |
| [17] |
Spurrier B, Ramalingam S, Nishizuka S. Reverse-phase protein lysate microarrays for cell signaling analysis[J]. Nat Protocols, 2008, 3(11): 1796-1808. DOI:10.1038/nprot.2008.179 |
| [18] |
Li J, Lu YL, Akbani R, et al. TCPA:a resource for cancer functional proteomics data[J]. Nat Methods, 2013, 10(11): 1046-1047. DOI:10.1038/nmeth.2650 |
| [19] |
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification[J]. Nat Biotechnol, 2008, 26(12): 1367-1372. DOI:10.1038/nbt.1511 |
| [20] |
Vasaikar SV, Straub P, Wang J, et al. LinkedOmics:analyzing multi-omics data within and across 32 cancer types[J]. Nucleic Acids Res, 2018, 46(D1): D956-963. DOI:10.1093/nar/gkx1090 |
| [21] |
Kaushik AK, Deberardinis RJ. Applications of metabolomics to study cancer metabolism[J]. Biochim Biophys Acta Rev Cancer, 2018, 1870(1): 2-14. DOI:10.1016/j.bbcan.2018.04.009 |
| [22] |
Beckonert O, Keun HC, Ebbels TMD, et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts[J]. Nat Protocols, 2007, 2(11): 2692-2703. DOI:10.1038/nprot.2007.376 |
| [23] |
Qiu YP, Cai GX, Zhou BS, et al. A distinct metabolic signature of human colorectal cancer with prognostic potential[J]. Clini Cancer Rese, 2014, 20(8): 2136-2146. DOI:10.1158/1078-0432.CCR-13-1939 |
| [24] |
Cavill R, Kamburov A, Ellis JK, et al. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells[J]. PLoS Comput Biol, 2011, 7(3): e1001113. DOI:10.1371/journal.pcbi.1001113 |
| [25] |
Tillinghast GW. Microarrays in the clinic[J]. Nat Biotechnol, 2010, 28(8): 810-812. DOI:10.1038/nbt0810-810 |
| [26] |
Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data[J]. Nat Rev Genet, 2010, 11(10): 733-739. DOI:10.1038/nrg2825 |
| [27] |
Robles AI, Harris CC. Integration of multiple "OMIC"biomarkers:A precision medicine strategy for lung cancer[J]. Lung Cancer, 2017, 107: 50-58. DOI:10.1016/j.lungcan.2016.06.003 |
| [28] |
TCGA GRCh38 metadata[M]. http://docs.cavatica.org/docs/tcga-grch38-metadata.
|
| [29] |
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours[J]. Nature, 2012, 490(7418): 61-70. DOI:10.1038/nature11412 |
| [30] |
Ebbels TMD, Cavill R. Bioinformatic methods in NMR-based metabolic profiling[J]. Progr Nucl Magnet Resonan Spectrosc, 2009, 55(4): 361-374. DOI:10.1016/j.pnmrs.2009.07.003 |
| [31] |
Fan TWM, Bandura LL, Higashi RM, et al. Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer cells[J]. Metabolomics, 2005, 1(4): 325-339. DOI:10.1007/s11306-005-0012-0 |
| [32] |
Nevedomskaya E, Perryman R, Solanki S, et al. A systems oncology approach identifies NT5E as a key metabolic regulator in tumor cells and modulator of platinum sensitivity[J]. J Proteome Res, 2016, 15(1): 280-290. DOI:10.1021/acs.jproteome.5b00793 |
| [33] |
Zhang G, He PJ, Tan H, et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer[J]. Clin Cancer Res, 2013, 19(18): 4983-4993. DOI:10.1158/1078-0432.CCR-13-0209 |
| [34] |
Wall JD, Tang LF, Zerbe B, et al. Estimating genotype error rates from high-coverage next-generation sequence data[J]. Genome Res, 2014, 24(11): 1734-1739. DOI:10.1101/gr.168393.113 |
| [35] |
Mathé E, Hays JL, Stover DG, et al. The omics revolution continues:the maturation of high-throughput biological data sources[J]. Yearb Med Inform, 2018, 27(1): 211-222. DOI:10.1055/s-0038-1667085 |