 2019, Vol. 40
2019, Vol. 40文章信息
- 高雪, 王慧, 王彤.
- Gao Xue, Wang Hui, Wang Tong.
- 孟德尔随机化中多效性偏倚校正方法简介
- Review on correction methods related to the pleiotropic effect in Mendelian randomization
- 中华流行病学杂志, 2019, 40(3): 360-365
- Chinese Journal of Epidemiology, 2019, 40(3): 360-365
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2019.03.020
-
文章历史
收稿日期: 2018-09-18




医学研究中,因果关联推断旨在对可控的暴露因素与结局之间的因果关系进行估计与评价,从而通过控制或干预暴露因素的水平改变相应结局。例如特定暴露对于人群发病的影响效应分析、特定药物或手术方式作用于疾病的疗效评估等。基于因果效应的分析与评价,对于确定疾病病因及干预方式、控制疾病进展或预后等方面均具有重要的指导意义。
工具变量模型最早见于计量经济学中,近年来被广泛应用于医学研究,用于推断暴露因素与结局之间的因果关联。具体而言,在经典回归模型中,解释变量与结果变量之间存在的未测量混杂因素以及逆向因果关联会导致解释变量与误差项相关,从而违背经典回归模型假设,产生偏倚的效应估计。而利用工具变量可排除解释变量中与误差项相关的部分,从而得到无偏倚的因果效应估计。在观察性研究中,工具变量可模拟随机对照研究中样本的随机化分配过程,从而实现混杂因素在暴露组间的可比性,使所研究结局的差异性由特定暴露进行解释,以此得到暴露因素与结局之间的因果关系[1]。
MR是以遗传变异作为工具变量的统计模型[1-2]。这一概念最初由Katan[3]于1986年提出,近年来被广泛应用于医学领域中。作为工具变量模型的一种,MR适用于基于观察性研究的因果效应推断;与传统的工具变量模型所不同的是,MR模型利用基因的遗传变异数据作为工具变量进行建模[4]。选择遗传变异作为工具变量的优势之处在于:个体的遗传变异先于疾病的结局,这样便排除了由于逆向因果问题所带来的混杂偏倚[5];同时,得益于生物信息技术的飞速发展,对遗传变异的测量能够达到很高的精度,因此相比于一般的工具变量模型,以遗传变异作为工具变量的MR模型也在很大程度上降低了由于测量误差所带来的估计偏倚[6]。
二、MR模型中工具变量的核心假设MR模型中,遗传变异作为工具变量需满足3个核心假设,当任意1个假设不满足时,该工具变量就是无效的(图 1):

|
| 注:Z:遗传变异工具变量;X:暴露因素;Y:结局;U:混杂因素 图 1 工具变量核心假设 |
关联性假设:遗传变异(Z)与暴露因素(X)之间存在稳健的强相关关系(γ≠0)。当关联性假设违背时,将会导致因果效应受弱工具变量的影响而产生偏倚[7-8]。如果能找到某个与暴露关联较强的遗传变异最好,但通常单个遗传变异对暴露因素的解释作用较弱,因此常选定多个与暴露因素相关的遗传变异作为工具变量,从而增大其对于暴露因素的解释强度,增加检验效能。
独立性假设:遗传变异(Z)与影响“暴露因素(X)—结局(Y)”关系的混杂因素(U)独立(φ1=0)。由于等位基因在配对形成过程中随机分离和组合,这一过程并不会受环境等后天因素的影响,遗传变异的这一特性保证了各个混杂因素在遗传变异不同亚组间的均衡性,为遗传变异与混杂因素的独立性假设提供了理论依据[9],但由于无法测量所有的混杂因素,因此这一假设是无法严格被证明的。
排他性假设:遗传变异只能通过暴露因素对结局产生作用,而不能通过其他路径影响结局(φ2=0)。事实上,遗传变异通常是通过复杂的生物学通路,而非只通过所研究的特定暴露因素影响结局[10]。现有的生物学研究还无法提供每种表型或疾病结局的完整遗传通路信息,因此无法严格证明遗传变异作为工具变量在通过暴露因素作用于结局外,是否还存在其他的通路影响结局,以及这些通路对于结局的影响作用大小。
三、遗传变异的多效性(pleiotropy effect,PE)及对MR的影响遗传变异的多效性是指遗传变异通过除特定“遗传变异—暴露因素—结局”路径之外的其他通路对结局产生影响[10-11]。当遗传变异存在多效性效应时,它可能通过影响“暴露因素—结局”关系的混杂因素作用于结局(导致独立性假设违背),也可能通过其他因素或直接作用于结局(导致排他性假设违背)。无论何种情况,该遗传变异作为工具变量都是无效的[12-13]。当无效工具变量引入模型后,若仍采用基于所有工具变量均有效的假设下的MR模型进行估计,必然会导致所研究的因果通路的效应估计出现偏倚,Ⅰ型错误率(假阳性率)增加[14-15]。
四、无效工具变量的多效性偏倚校正方法遗传变异的多效性效应是普遍存在的,而其对于MR模型的影响又是不可忽略的,因此如何排除由于遗传变异多效性效应导致的工具变量失效对于待研究的因果效应通路的偏倚,是一个亟待解决的问题,也是该领域中的重要研究内容[2]。现有研究中提出了诸多解决无效工具变量问题的方法及模型,现分别从工具变量筛选、无效工具变量检验、基于汇总数据的多效性校正模型构建、基于个体数据的多效性校正模型以及敏感性分析5个方面详细阐述无效工具变量的多效性偏倚校正方法。
1.工具变量筛选:针对无效工具变量问题,在利用MR进行建模之前,应当有针对性地选取工具变量[16]。例如待研究的暴露因素是可溶性蛋白质标记物,那么通常会有一个基因片段直接编码该蛋白质(例如,C-反应蛋白基因片段编码C-反应蛋白[17]),或调节蛋白质表达[例如,白细胞介素-6(IL-6)受体调控IL-6表达[18]]。利用这些基因片段上的遗传变异作为工具变量通常是一种比较理想的情况。然而,诸多涉及多因子、多基因的复杂性状,其表达过程包含多个基因上的片段参与。针对这种复杂性状与结局之间的分析,则需要选定不同基因片段上的遗传变异作为工具变量,才能保证其与相应性状稳健的强相关性[19-20]。然而,不同基因区域上的遗传变异则有很大的可能存在多效性,即不同的基因片段通过不同的生物学通路影响结局[21]。
另有研究提出在选取工具变量时排除可能存在多效性的遗传变异[22-23]。这在一定程度上可避免由于工具变量多效性所导致的结果偏倚,但一旦为了避免可能的多效性而去掉对暴露因素具有强解释作用的遗传变异,又会导致工具变量关联强度的损失,产生弱工具变量问题。并且,在对遗传变异的生物学信息没有充分的了解时,如何识别出无效工具变量本身就是一个困难的工作。因此单纯通过建模前期的工具变量选择,无法确保可以完全避免无效工具变量对于结果的影响,应进一步进行工具变量有效性检验、优化模型建立以及敏感性分析等,以检验并校正无效工具变量的多效性引入的偏倚。
2.无效工具变量检验:当多个工具变量纳入模型时,针对个体数据可利用Sargan检验,针对汇总数据可利用Cochran’s Q检验、I2统计量、似然比检验等评价工具变量的整体有效性[8, 24-26]。在经济学模型中,这种检验方法称为过度识别检验,其原假设为所有有效工具变量确定相同的因果效应[27-28]。检验以各个工具变量得到的因果效应值的异质性作为统计量,其基本思想为若每个工具变量的因果效应估计值是相似的,即异质性不显著,此时不拒绝原假设,从而证明了工具变量的整体有效性;反之,如果由不同工具变量得到的因果效应存在显著的异质性,此时拒绝原假设,说明存在无效工具变量,这些无效工具变量的因果效应估计值与其他工具变量估计值存在显著的差异。
下面通过公式具体说明无效工具变量的异质性原理:
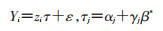 (1)
(1)
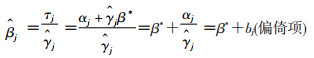 (2)
(2)
由图 2及公式1可得,“工具变量—结局”关联系数τj等于工具变量多效性αj加上“工具变量—暴露因素”关联系数γj与“暴露因素—结局”之间的因果关联系数β*的乘积。由公式2可得,单个工具变量的比例估计值为“工具变量—结局”关联系数τj除以“工具变量—暴露因素”关联系数γj。在工具变量均有效的前提假设下(αj=0),各个遗传变异的比例估计值即为真实的因果效应值β*;而在工具变量因存在多效性而成为无效工具变量时(αj≠0),比例估计值为真实的因果效应值β*加上由多效性所引入的偏倚项bj。在进行正式的统计检验前,可以利用“工具变量—结局”与“工具变量—暴露因素”之间的关联系数(τj,γj)描绘散点图,由此将各个工具变量的有效性信息进行可视化分析。由上述结论可得,有效工具变量所表示的点趋近以真实的因果效应值为斜率的直线,而无效工具变量所表示的点则背离直线[21]。

|
| 注:Z:遗传变异工具变量;X:暴露因素;Y:结局 图 2 工具变量—暴露因素—结局关联关系图 |
3.基于汇总数据的多效性校正模型简述:汇总数据是指从全基因组关联研究等获取的遗传变异与相关表型及结局的关联关系的参数估计量。针对汇总数据的多效性校正模型,主要介绍多变量MR方法、基于中位数及众数的方法、MR-Egger回归方法。
(1)多变量MR方法:Burgess和Thompson[29]于2015年提出多变量MR模型,旨在校正与工具变量相关的可测量混杂因素对于因果效应的影响,从而排除多效性偏倚。多变量MR方法利用二阶段回归的思想,第一阶段先将“工具变量—结局”关联系数与“工具变量—混杂因素”关联系数进行回归,目的在于从总体效应中排除工具变量对混杂因素的多效性效应;第二阶段再利用第一阶段的回归残差与“工具变量—暴露因素”关联系数进行回归[23],以此得出暴露因素与结局的因果关联。
该方法类似于同时估计多个干预效应的析因设计随机化试验,其优点在于可以同时分析多个潜在的暴露因素与结局的因果效应,但缺陷在于该方法仅能校正少数已知通路的影响,而对于工具变量到结局的未知通路仍然无法进行校正。从遗传变异的多效性效应角度来说,该方法仅能排除已知多效性,而无法排除未知多效性对于估计系数的偏倚影响。
(2)基于中位数的方法:单个遗传变异下的因果效应的比例估计(ratio estimate)为“工具变量—结局”关联系数与“工具变量—暴露因素”关联系数的比值[9]。Bowden等[30]在2015年提出加权中位数估计(WME)和惩罚加权中位数估计(PWME)方法。WME首先利用delta方法计算每个工具变量的比例估计值的权重、对应的百分位数以及总体经验分布函数,再取该经验分布函数下的中位数作为因果效应估计值。WME在50%以上的权重来自于有效工具变量时即可得到因果效应的一致性估计;PWME首先对存在异质性的比例估计值对应的权重进行惩罚,再利用惩罚后的权重构造分布函数并估计因果效应。相比于WME,PWME进一步减弱了由于遗传变异多效性而导致的因果效应估计值之间的异质性对于总体因果效应估计的影响[30]。
(3)基于众数的方法(MBE):Hartwig等[31]于2017年提出MBE。该方法基于众数的思想,利用各个工具变量的比例估计值构造平滑的核密度函数[32](类似于由频数直方图中代表每组频数的长方形顶端中点构成一条平滑的曲线),并取核密度函数最大值点对应的参数值作为因果效应估计值。考虑到估计精度对于结果的影响,可以将每个工具变量的比例估计值的方差倒数作为权重并带入经验核密度函数。MBE得到一致估计的前提假设为在所有工具变量产生的偏倚项的不同取值中,偏倚项为0(即不存在多效性)的项数最多(简单多数原则)。这个假设相比于WME方法中过半数有效工具变量的假设更加宽松,因而在实际问题中更具普遍性。
(4)MR-Egger回归方法:Bowden等[33]于2015年提出MR-Egger方法。在满足工具变量多效性效应与工具变量对暴露因素的影响效应相互独立的假设(InSIDE)时,借鉴Egger回归思想,将工具变量多效性偏倚类比于Meta分析中的小样本偏倚[34],利用Meta分析中小样本偏倚的检验方法进行检验并校正由工具变量多效性产生的偏倚。相比于基于中位数及众数的估计,该方法能够有效识别工具变量的整体多效性,并且即使在全部工具变量均存在多效性的情况下仍可得到一致的估计。然而,该方法对假设仍有比较高的敏感性,当Inside假设不满足时,Egger回归得到的估计量同样会受到偏倚影响。
在进行MR-Egger回归之前,可以参考Meta分析方法,构建基于各个工具变量强度与因果效应值的漏斗图,初步判定工具变量是否存在多效性[33, 35]。若遗传变异不存在多效性,那么从漏斗图上看,各个遗传变异所代表的点应该是对称分布的;反之,漏斗图上任何形式的不对称性都提示工具变量多效性的存在,应进一步利用MR-Egger回归校正其对于整体因果效应的影响。
4.基于个体数据的多效性校正模型:当研究者可以获得样本中每个个体的遗传变异及相关表型数据时,可以利用基于个体数据的MR模型进行因果效应估计。针对基于个体数据的多效性校正模型,主要介绍sisVIVE方法、结合两阶段硬阈值(two- stage hard thresholding,TSHT)、多效性-稳健MR模型(PRMR)。
(1)sisVIVE:Kang等[13]于2016年提出sisVIVE估计量。该方法借用Lasso惩罚回归方法的思想,在传统的两阶段最小二乘模型(TSLS)的基础之上将多效性效应进行基于L1范数的惩罚,从而筛选无效工具变量并校正其多效性引入的估计偏倚。该方法类似于Gautier等[36]、Belloni等[37]分别于2011年及2012年提出的方法,但这两种方法需要建模前明确定义无效工具变量才可获得效应的估计;另外,Liao[38]、Cheng和Liao[39]也提出基于压缩估计的方法确定无效工具变量,但这两种方法需要事先确定一个有效工具变量的子集。相比较而言,sisVIVE估计量的明显优势在于仅依靠无效工具变量所占比例低于50%的前提假设(过半数原则),而不需要提供具体哪些遗传变异是有效工具变量的先验信息,便可得到一致的估计量,这在很大程度上放宽了模型的限制条件。
(2)TSHT投票法:Guo等[12]于2017年提出TSHT投票法和简单多数投票的估计方法。两阶段硬阈值法的第一阶段筛选出与暴露因素具有强相关的工具变量集合S[40],第二阶段从集合S中筛选出有效的工具变量集合P|j|。经历了TSHT的两个步骤后,可以确定|S|个集合V|j|=S∩P|j|(|S|为集合的势,对于有限集合,即代表集合中元素的个数)。之后利用简单多数投票原则合并V|j|,取得最终的工具变量集合,该集合中的每个遗传变异均满足工具变量的3个核心假设。最后利用该集合中的工具变量进行建模从而获得一致的因果效应估计。
相比于sisVIVE的有效工具变量过半数假设,TSHT投票法所依赖的有效工具变量占多数假设更为宽松;但该方法直接剔除无效工具变量,而非校正其多效性效应,若将对暴露因素具有强代表性的工具变量剔除后,剩余工具变量对暴露因素的代表性不足将会同样引起估计的偏倚,即弱工具变量偏倚。
(3)PRMR:van Kippersluis和Rietveld[41]于2017年提出PRMR。该模型结合辅助回归与合理外生方法,首先在工具变量对暴露因素效应为零的子样本中,利用辅助回归估计多效性效应;之后假定遗传变异在各个子样本中的效应同质,并将由辅助回归得到的多效性效应估计值纳入合理外生模型进行校正,从而得到无偏的因果效应[42]。在实际研究中,是否可以找到工具变量对暴露因素无关联效应的子样本是PRMR能否得以有效运用的关键。也就是说,只有存在满足上述条件的子样本,才能通过该方法进行有效的估计。
5.敏感性分析:当遗传变异作为工具变量的假设缺乏可靠的生物学依据时,任何不考虑到工具变量假设违背情况而忽略对结果进行稳健性评价,由此得到的结论都是不完整并且值得怀疑的[43]。因此,有必要通过敏感性分析评价结果对于假设成立证据不充分甚至违背情况下的敏感程度。实证分析中,常利用不同的多效性效应校正模型对数据进行分析,并比较各种方法所得结果的差异性,从而评估模型的稳健性。另外,也可以根据遗传变异的生物学信息对工具变量进行分组,并比较不同组合建模估计所得因果效应的异质性[16]。不同的工具变量及组合得到的因果效应若一致,则说明结果较为稳健,反之则说明现有模型对于无效工具变量假设较为敏感,有必要更进一步进行分析。针对MR模型的敏感性分析方法还有系统去变量法[44]、证伪检验[45]等,在此不一一赘述。
五、总结基于工具变量的MR模型,因其可以控制混杂因素、排除逆向因果问题,在因果关联推断中得到了广泛应用。工具变量的核心假设违背可以归结为弱工具变量以及无效工具变量问题。遗传变异与暴露因素的关联通常可以通过研究者实际掌握的样本数据进行估计,因此是否存在弱工具变量问题是较容易评价的;而基于对生物学信息研究的局限性,遗传变异是否存在多效性问题是难以准确评价的,因此其作为工具变量的有效性则难以被严格验证。针对这一问题,本文从工具变量选择、无效工具变量检验、稳健模型的构建及敏感性分析等方面阐述了如何识别及校正无效工具变量的多效性效应对于因果效应估计的偏倚影响。
在稳健模型的构建上,本文分别介绍了基于汇总数据及个体数据的多效性校正模型。需要注意的是,在针对实际问题的分析中,不存在适用于所有实证背景的方法,应该结合数据类型、工具变量个数以及遗传变异的生物学信息等多方面进行参考,从而选用适合的模型进行分析。另外,除了应用稳健的方法外,还有一些问题需要注意。例如检验效能的保证[46-47],样本含量的确定及样本数据的收集[4, 48],暴露因素与结局的正确定义[21]、选择偏倚问题[49-50]、人群分层问题[51]等。因此在利用MR模型进行推断因果效应时,除了对工具变量以及模型的恰当选择外,针对这些问题也应该做出恰当的考虑和处理,以保证对于因果效应的准确估计和评价。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
叶冬青, 王彤, 王鸣, 等. 流行病学进展(第13卷)[M]. 北京: 人民卫生出版社, 2017. Ye DQ, Wang T, Wang M, et al. Progress in Epidemiology(thirteenth volume)[M]. Beijing: People's Medical Publishing House, 2017. |
| [2] |
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization[J]. JAMA, 2017, 318(19): 1925-1926. DOI:10.1001/jama.2017.17219 |
| [3] |
Katan MB. Commentary:Mendelian randomization, 18 years on[J]. Int J Epidemiol, 2004, 33(1): 10-11. DOI:10.1093/ije/dyh023 |
| [4] |
Smith GD, Ebrahim S. 'Mendelian randomization':can genetic epidemiology contribute to understanding environmental determinants of disease?[J]. Int J Epidemiol, 2003, 32(1): 1-22. DOI:10.1093/ije/dyg070 |
| [5] |
Smith GD, Lawlor DA, Harbord R, et al. Clustered environments and randomized genes:a fundamental distinction between conventional and genetic epidemiology[J]. PLoS Med, 2007, 4(12): e352. DOI:10.1371/journal.pmed.0040352 |
| [6] |
Evans DM, Davey Smith G. Mendelian randomization:new applications in the coming age of hypothesis-free causality[J]. Annu Rev Genomics Hum Genet, 2015, 16: 327-350. DOI:10.1146/annurev-genom-090314-050016 |
| [7] |
Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies[J]. Int J Epidemiol, 2011, 40(3): 755-764. DOI:10.1093/ije/dyr036 |
| [8] |
Davies NM, von Hinke Kessler Scholder S, Farbmacher H, et al. The many weak instruments problem and Mendelian randomization[J]. Stat Med, 2015, 34(3): 454-468. DOI:10.1002/sim.6358 |
| [9] |
Lawlor DA, Harbord RM, Sterne JAC, et al. Mendelian randomization:using genes as instruments for making causal inferences in epidemiology[J]. Stat Med, 2008, 27(8): 1133-1163. DOI:10.1002/sim.3034 |
| [10] |
Hackinger S, Zeggini E. Statistical methods to detect pleiotropy in human complex traits[J]. Open Biol, 2017, 7(11): 170125. DOI:10.1098/rsob.170125 |
| [11] |
Wagner GP, Zhang JZ. The pleiotropic structure of the genotype-phenotype map:the evolvability of complex organisms[J]. Nat Rev Genet, 2011, 12(3): 204-213. DOI:10.1038/nrg2949 |
| [12] |
Guo ZJ, Kang H, Tony CT, et al. Confidence intervals for causal effects with invalid instruments by using two-stage hard thresholding with voting[J]. J Roy Statist Soc Ser B, 2018, 80(4): 793-815. DOI:10.1111/rssb.12275 |
| [13] |
Kang H, Zhang AR, Cai TT, et al. Instrumental variables estimation with some invalid instruments and its application to Mendelian randomization[J]. J Am Stat Assoc, 2016, 111(513): 132-144. DOI:10.1080/01621459.2014.994705 |
| [14] |
Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization[J]. Int J Epidemiol, 2013, 42(4): 1134-1144. DOI:10.1093/ije/dyt093 |
| [15] |
Pickrell J. Fulfilling the Promise of Mendelian Randomization[M]. bioRxiv, 2015. DOI:10.1101/018150
|
| [16] |
Emdin CA, Khera AV, Natarajan P, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease[J]. JAMA, 2017, 317(6): 626-634. DOI:10.1001/jama.2016.21042 |
| [17] |
C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC). Association between C reactive protein and coronary heart disease:Mendelian randomisation analysis based on individual participant data[J]. BMJ, 2011, 342: d548. DOI:10.1136/bmj.d548 |
| [18] |
IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease:a collaborative Meta-analysis of 82 studies[J]. Lancet, 2012, 379(9822): 1205-1213. DOI:10.1016/S0140-6736(11)61931-4 |
| [19] |
Hägg S, Fall T, Ploner A, et al. Adiposity as a cause of cardiovascular disease:a Mendelian randomization study[J]. Int J Epidemiol, 2015, 44(2): 578-586. DOI:10.1093/ije/dyv094 |
| [20] |
The International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk[J]. Nature, 2011, 478(7367): 103-109. DOI:10.1038/nature10405 |
| [21] |
Burgess S, Bowden J, Fall T, et al. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants[J]. Epidemiology, 2017, 28(1): 30-42. DOI:10.1097/EDE.0000000000000559 |
| [22] |
Mokry LE, Ahmad O, Forgetta V, et al. Mendelian randomisation applied to drug development in cardiovascular disease:a review[J]. J Med Genet, 2015, 52(2): 71-79. DOI:10.1136/jmedgenet-2014-102438 |
| [23] |
Burgess S, Freitag DF, Khan H, et al. Using multivariable Mendelian randomization to disentangle the causal effects of lipid fractions[J]. PLoS One, 2014, 9(10): e108891. DOI:10.1371/journal.pone.0108891 |
| [24] |
Palmer TM, Lawlor DA, Harbord RM, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors[J]. Stat Methods Med Res, 2012, 21(3): 223-242. DOI:10.1177/0962280210394459 |
| [25] |
del Greco MF, Minelli C, Sheehan NA, et al. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome[J]. Stat Med, 2015, 34(21): 2926-2940. DOI:10.1002/sim.6522 |
| [26] |
Higgins JP, Thompson SG, Deeks JJ, et al. Measuring inconsistency in Meta-analyses[J]. BMJ, 2003, 327(7414): 557-560. DOI:10.1136/bmj.327.7414.557 |
| [27] |
Wooldridge JM. Introductory Econometrics:a Modern Approach[M]. 5th ed. Mason, OH: South-Western, 2013.
|
| [28] |
陈强. 高级计量经济学及Stata应用[M]. 北京: 高等教育出版社, 2010. Chen Q. Advanced Econometrics and Applications of Stata[M]. Beijing: Higher Education Press, 2010. |
| [29] |
Burgess S, Thompson SG. Multivariable Mendelian randomization:the use of pleiotropic genetic variants to estimate causal effects[J]. Am J Epidemiol, 2015, 181(4): 251-260. DOI:10.1093/aje/kwu283 |
| [30] |
Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator[J]. Genet Epidemiol, 2016, 40(4): 304-314. DOI:10.1002/gepi.21965 |
| [31] |
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption[J]. Int J Epidemiol, 2017, 46(6): 1985-1998. DOI:10.1093/ije/dyx102 |
| [32] |
Bickel DR, Frühwirth R. On a fast, robust estimator of the mode:comparisons to other robust estimators with applications[J]. Comput Statist Data Anal, 2006, 50(12): 3500-3530. DOI:10.1016/j.csda.2005.07.011 |
| [33] |
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments:effect estimation and bias detection through Egger regression[J]. Int J Epidemiol, 2015, 44(2): 512-525. DOI:10.1093/ije/dyv080 |
| [34] |
Egger M, Davey Smith G, Schneider M, et al. Bias in Meta-analysis detected by a simple, graphical test[J]. BMJ, 1997, 315(7109): 629-634. DOI:10.1136/bmj.315.7109.629 |
| [35] |
Sterne JAC, Sutton AJ, Ioannidis JPA, et al. Recommendations for examining and interpreting funnel plot asymmetry in Meta-analyses of randomised controlled trials[J]. BMJ, 2011, 343: d4002. DOI:10.1136/bmj.d4002 |
| [36] |
Gautier E, Tsybakov A, Rose C. High-dimensional Instrumental Variables Regression and Confidence Sets[M]. arXiv, 2011.
|
| [37] |
Belloni A, Chen D, Chernozhukov V, et al. Sparse models and methods for optimal instruments with an application to eminent domain[J]. Econometrica, 2012, 80(6): 2369-2429. DOI:10.3982/ECTA9626 |
| [38] |
Liao ZP. Adaptive GMM shrinkage estimation with consistent moment selection[J]. Econometric Theory, 2013, 29(5): 857-904. DOI:10.1017/S0266466612000783 |
| [39] |
Cheng X, Liao ZP. Select the valid and relevant moments:an information-based LASSO for GMM with many moments[J]. J Econometrics, 2015, 186(2): 443-464. DOI:10.1016/j.jeconom.2015.02.019 |
| [40] |
Donoho DL. De-noising by soft-thresholding[J]. IEEE Trans Inf Theory, 1995, 41(3): 613-627. DOI:10.1109/18.382009 |
| [41] |
van Kippersluis H, Rietveld CA. Pleiotropy-robust Mendelian randomization[J]. Int J Epidemiol, 2018, 47(4): 1279-1288. DOI:10.1093/ije/dyx002 |
| [42] |
Conley TG, Hansen CB, Rossi PE. Plausibly exogenous[J]. Rev Econ Statist, 2012, 94(1): 260-272. DOI:10.1162/REST_a_00139 |
| [43] |
Swanson SA, Hernán MA. Commentary:how to report instrumental variable analyses (suggestions welcome)[J]. Epidemiology, 2013, 24(3): 370-374. DOI:10.1097/EDE.0b013e31828d0590 |
| [44] |
Smith JG, Luk K, Schulz CA, et al. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis[J]. JAMA, 2014, 312(17): 1764-1771. DOI:10.1001/jama.2014.13959 |
| [45] |
Alsan M. The effect of the TseTse fly on African development[J]. Am Econ Rev, 2015, 105(1): 382-410. DOI:10.1257/aer.20130604 |
| [46] |
Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies[J]. Int J Epidemiol, 2013, 42(5): 1497-1501. DOI:10.1093/ije/dyt179 |
| [47] |
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants[J]. Int J Epidemiol, 2011, 40(3): 740-752. DOI:10.1093/ije/dyq151 |
| [48] |
Freeman G, Cowling BJ, Schooling CM. Power and sample size calculations for Mendelian randomization studies using one genetic instrument[J]. Int J Epidemiol, 2013, 42(4): 1157-1163. DOI:10.1093/ije/dyt110 |
| [49] |
Swanson SA, Robins JM, Miller M, et al. Selecting on treatment:a pervasive form of bias in instrumental variable analyses[J]. Am J Epidemiol, 2015, 181(3): 191-197. DOI:10.1093/aje/kwu284 |
| [50] |
Ertefaie A, Small D, Flory J, et al. A sensitivity analysis to assess bias due to selecting subjects based on treatment received[J]. Epidemiology, 2016, 27(2): e5-7. DOI:10.1097/EDE.0000000000000430 |
| [51] |
Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies[J]. Nat Genet, 2006, 38(8): 904-909. DOI:10.1038/ng1847 |