 2019, Vol. 40
2019, Vol. 40文章信息
- 王译葵, 陈晓晨, 王继宝, 段星, 周素娟, 杨锦, 杨涛, 叶润华, 杨跃诚, 姚仕堂, 段松, 何纳.
- Wang Yikui, Chen Xiaochen, Wang Jibao, Duan Xing, Zhou Sujuan, Yang Jin, Yang Tao, Ye Runhua, Yang Yuecheng, Yao Shitang, Duan Song, He Na.
- 德宏傣族景颇族自治州2016年HIV/HCV合并感染者HCV分子传播簇分析
- Molecular transmission clusters on HCV genotypes among newly reported HIV/HCV co-infection in Dehong Dai and Jingpo autonomous prefecture of Yunnan province, 2016
- 中华流行病学杂志, 2019, 40(2): 191-195
- Chinese Journal of Epidemiology, 2019, 40(2): 191-195
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2019.02.013
-
文章历史
收稿日期: 2018-08-14




2. 复旦大学公共卫生学院流行病学教研室 公共卫生安全教育部重点实验室, 上海 200032
2. Department of Epidemiology, School of Public Health, The Key Laboratory for Public Health Safety of Ministry of Education, Fudan University, Shanghai 200032, China
艾滋病等传染病的传播网络应用,从之前的单纯依靠问卷或者同伴追踪等方法构建的社会或性传播网络[1-2],到后来的借助HIV序列的遗传信息建立的分子传播网络[3],尝试尽可能还原宏观的感染者社会网络,快捷高效[4],目前多用于验证现场流行病学调查的结果和结论。近年来,国际上已将该方法应用到与HIV有着相似传播途径的HCV研究[5]。然而国内尚无基于进化分析的HCV分子传播网络研究的报道。本研究分析德宏傣族景颇族自治州(德宏州)HIV/HCV合并感染者中HCV主要毒株亚型的流行分布特征,探讨HCV主要亚型分子传播簇及其影响因素。
对象与方法本研究的基因距离阈值为3%,即基因距离≤0.03,分别对HCV的3b、6n、6u、1a和3a亚型进行系统进化树的构建(依据NS5B基因扩增区)。
1.研究对象:利用全国艾滋病综合防治数据信息系统下载德宏州2016年1月1日至12月31日新报告HIV/AIDS病例数据共1 112例,收集其社会人口学相关信息。采集能满足HCV检测要求的共1 045份样本,通过两种不同ELISA法筛选检出HCV抗体阳性样本336份,筛选出血浆量≥200 μl的318份样本。
2.实验方法:
(1)HCV抗体检测:初筛试剂使用厦门英科新创科技有限公司生产的HCV抗体诊断试剂盒,对2016年德宏州新报告HIV感染者进行HCV抗体初筛;复检试剂使用珠海丽珠试剂股份有限公司生产的HCV抗体诊断试剂盒,严格按照试剂盒说明书要求操作,经两种不同ELISA检测HCV抗体,均为阳性的样本进行核酸扩增。
(2)HCV核酸提取和扩增:参照QIAamp Viral RNA Mini Kit试剂盒(德国Qiagen公司)说明书,从140 μl血浆中提取HCV RNA,立刻进行HCV核酸扩增。参照SuperScript Ⅲ First-Strand Synthesis System for RT-PCR试剂盒(美国Invitrogen公司)说明书进行操作,以20 μl HCV RNA为模板,采用随机引物合成cDNA。分别使用TaKaRa Ex Taq试剂[宝生物工程(大连)有限公司]和SYBR®Premix Ex TaqTM Ⅱ对HCV的CE1和NS5B基因片段进行巢式PCR扩增,将第2轮PCR产物经质量分数为1%的琼脂糖凝胶电泳鉴定,扩增成功的PCR产物送北京博迈德基因技术有限公司测序。所用扩增引物和测序引物是参照Roffi等[6]研究选择,见表 1。其中CE1区测序引物为CE1-ISP和CE1-IAP,NS5B区测序引物为NS5S2和NS5A2。

3.基因型分析:使用ChromasPro1.5和BioEdit 7.0.9.0软件,参照国际参考株的顺序[7],对样本序列拼接、校对、比对;将序列提交到HCV数据库(http://hcv.lanl.gov/content/index),进行亚型的初步判定。应用Mega 7.0软件的Neighbor-joining方法建立系统进化树,Bootstrap重复检测值设为1 000次,根据系统进化树进一步确定HCV基因亚型。同一样本两个基因片段亚型不一致的使用SimPlot 3.5.1软件进行重组分析。
4.构建HCV分子传播网络:用Mega 7.0软件计算传播簇内个体之间的两两基因距离,Boot strap设置1 000,选择tamura-Nei参数模型(TN93模型)。本研究中将基因距离阈值为3%即基因距离≤0.03和进化树节点值≥95%,并且构建系统进化树,簇内序列数≥2条即认为传播簇。而成簇率表示进入簇内的序列数占总序列数的百分比,某人群的成簇率越高,其传播风险越高。序列间基因距离小于阈值则表示他们的感染密切相关,可能存在传播关系,在网络中会被连接在一起,即分子传播网络。利用Gephi 0.9.2软件实现传播网络的可视化。有两个重要概念即节点和边。前者代表一条HCV序列或者一个HCV感染者,后者代表连着的两个HCV感染者存在潜在的传播关系。在网络中节点与节点之间相互连接,而一个节点连接其他节点的边的数目则称为度值,可反映出每个节点对网络结构的重要性[8]。依据NS5B基因扩增区,本研究设定基因距离阈值为3%(即基因距离≤0.03),分别对HCV基因亚型系统进化树的构建。
5.统计学分析:应用SPSS 22.0软件对数据进行统计分析。计数资料采用例数与构成比(%)描述。不同组间差异采用χ2检验或者Fisher确切概率法分析。双侧检验,以P<0.05为差异有统计学意义。
结果1. HIV/HCV合并感染者基本特征及HCV基因亚型:2016年德宏州HIV/HCV合并感染者336例,筛选出HIV/HCV合并感染且血浆量≥200 μl的318份样本,满足HCV基因亚型检测要求共267份样本。以男性(93.3%)、≥25岁(79.8%)、景颇族(39.7%)、小学及以下文化程度(76.8%)和缅甸籍(80.9%)为主。HCV基因亚型呈多样化,主要有3b、6n、6u、1a、3a和其他亚型,构成比分别为32.6%(87/267)、18.4%(49/267)、15.7%(42/267)、13.1%(35/267)、11.2%(30/267)和9.0%(24/267)。除性别、民族在不同HCV基因亚型的分布差异有统计学意义外,其他特征差异无统计学意义(P>0.05)。见表 2。

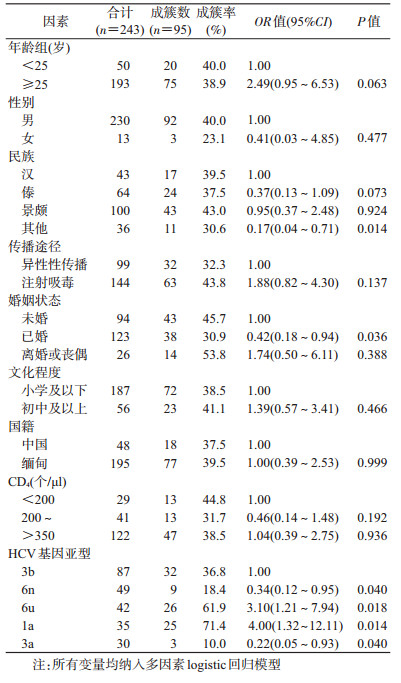
2.不同亚型的成簇率及总体成簇影响因素:共有243例样本,总成簇率为39.1%(95/243)。3b、6n、6u、1a和3a亚型的成簇率分别是36.8%(32/87)、18.4%(9/49)、61.9%(26/42)、71.4%(25/35)和10.0%(3/30)。多因素logistic回归分析结果显示,除傣族和景颇族外的其他民族(与汉族相比,OR=0.17,95%CI:0.04~0.71)、已婚(与未婚相比,OR=0.42,95%CI:0.18~0.94)、6n与3a亚型(与3b亚型相比,OR=0.34,95%CI:0.12~0.95;OR=0.22,95%CI:0.05~0.93)较难形成传播簇;6u和1a亚型(与3b亚型相比,OR=3.10,95%CI:1.21~7.94;OR=4.00,95%CI:1.32~12.11)较易形成传播簇。见表 3。
3. HCV基因亚型构建的分子传播簇网络:HIV/HCV合并感染者的民族、婚姻状况、HCV基因亚型是HCV分子传播簇形成的关联因素。根据HCV主要基因亚型构建分子传播簇网络,1a、3b和6u亚型形成的分子网络图更为复杂,每个节点连接的边较多,代表传播关系更复杂,防控难度更大(见图 1)。传播簇内≥10条边的有23例,其中,3b、6u、1a亚型分别有3、11、9例,且均为男性,19例缅甸籍(82.6%),16例(69.6%)为注射吸毒传播,21例为少数民族。

|
| 图 1 2016年德宏州HIV/HCV合并感染者HCV基因亚型构建的分子传播簇网络 |
本研究发现,德宏州新报告HIV/HCV合并感染者中HCV基因亚型呈多样化,主要有3b、6n、6u、1a和3a等亚型。已有研究表明,静脉吸毒人群的HCV基因亚型主要为3型和6型[9]。本研究对象多为注射吸毒感染,主要流行毒株和优势毒株也是3型与6型,说明HCV基因亚型与感染途径确实存在着较高的相关性。德宏州与我国其他地区的1a、2b型为主要优势亚型不同[7, 10-11]。德宏州地处我国西南边陲、毗邻缅甸,以6型、3型和1型为主要基因亚型[12-13]。
多因素logistic回归分析结果发现,参与成簇的HIV/HCV合并感染者占39.1%,高于其他研究[5, 14]。其中,1a亚型的成簇率最高(64.1%),6u(61.9%),提示亚型1a、6u传播风险高于其他亚型。多因素logistic回归分析结果显示,民族、婚姻状况、基因亚型与HIV分子成簇率有显著关联,是传播风险的主要影响因素。已婚的成簇率低于单身,而且发生成簇的危险性明显低于单身,可能与已婚的感染者具有家庭责任感有关,也会受到一定的约束,发生聚集注射吸毒等高危行为的可能性较小。本研究发现,注射吸毒的男性更容易形成单个节点连接较多边的传播簇,说明传播关系更为复杂。在今后的防控工作中,应该对此类人群重点关注。另外,基因距离阈值的设定目前并没有统一的标准,构建分子传播网络的方法依据研究的样本特征和目的来选择[4]。如前瞻性研究为了发现更多的新发感染者和高危传播者,可以适当放宽阈值。HIV分子网络基因距离阈值一般为1.0%~4.5%。而现有的国际上有关HCV分子网络研究其基因距离阈值也设置为3%[5],故本研究也将基因距离阈值设定为3%。
本研究存在不足。一是检测HCV抗体阳性仅能判断HCV感染状态,不能判断HCV感染的具体时间,而感染时间的长短可能对病毒变异有影响。有研究提示尽管HCV的基因变化速率较快,但在个体水平上的变异却有限,HCV在同一个体中短时间内变异不大[15]。HCV感染者整体呈基因离散率(基因距离)随着流行时间的延长而增加,HCV基因亚型的分子传播簇总数可能造成一定程度上的低估。需进一步的前瞻性探讨HCV感染时间长短对其分子传播簇网络是否有影响。二是研究对象有我国和缅甸两国人群,由于中国籍样本量太少,未按国籍讨论,德宏州HIV/HCV合并感染者的民族、婚姻状况、HCV基因亚型是HCV分子传播簇形成的关联因素。利用基因亚型分析特征构建的传播网络,可以最大限度的早发现作为传染源的HCV感染者,帮助我们识别那些传播风险最大的个体,为当地预测估计HIV感染者中HCV传播流行趋势和制定针对性干预策略提供直接的科学依据,具有重大公共卫生学意义。
利益冲突 所有作者均声明不存在利益冲突
| [1] |
Hellard M, Rolls DA, Sacks-Davis R, et al. The impact of injecting networks on hepatitis C transmission and treatment in people who inject drugs[J]. Hepatology, 2014, 60(6): 1861-1870. DOI:10.1002/hep.27403 |
| [2] |
Robinson K, Cohen T, Colijn C. The dynamics of sexual contact networks:effects on disease spread and control[J]. Theor Popul Biol, 2012, 81(2): 89-96. DOI:10.1016/j.tpb.2011.12.009 |
| [3] |
Lewis F, Hughes GJ, Rambaut A, et al. Episodic sexual transmission of HIV revealed by molecular phylodynamics[J]. PLoS Med, 2008, 5(3): e50. DOI:10.1371/journal.pmed.0050050 |
| [4] |
黎裕雪云, 冯毅, 阮玉华, 等. HIV分子传播网络研究进展[J]. 中国艾滋病性病, 2016, 22(11): 940-942. Li YXY, Feng Y, Ruan YH, et al. Advance of HIV molecular transmission network[J]. Chin J AIDS STD, 2016, 22(11): 940-942. DOI:10.13419/j.cnki.aids.2016.11.29 |
| [5] |
Bartlett SR, Wertheim JO, Bull RA, et al. A molecular transmission network of recent hepatitis C infection in people with and without HIV:implications for targeted treatment strategies[J]. J Viral Hepat, 2017, 24(5): 404-411. DOI:10.1111/jvh.12652 |
| [6] |
Roffi L, Redaelli A, Colloredo G, et al. Outcome of liver disease in a large cohort of histologically proven chronic hepatitis C:influence of HCV genotype[J]. Eur J Gastroenterol Hepatol, 2001, 13(5): 501-506. DOI:10.1097/00042737-200105000-00007 |
| [7] |
Zhang TJ, Tully DC, Zhou SJ, et al. Characteristics of HCV co-infection among HIV infected individuals from an area with high risk of blood-borne infections in central China[J]. PLoS One, 2014, 9(4): e94219. DOI:10.1371/journal.pone.0094219 |
| [8] |
Helleringer S, Kohler HP. Sexual network structure and the spread of HIV in Africa:evidence from Likoma Island, Malawi[J]. AIDS, 2007, 21(17): 2323-2332. DOI:10.1097/QAD.0b013e328285df98 |
| [9] |
Garten RJ, Zhang J, Lai S, et al. Coinfection with HIV and hepatitis C virus among injection drug users in southern China[J]. Clin Infect Dis, 2005, 41. DOI:10.1086/429491 |
| [10] |
Zhang LQ, Chen ZW, Cao YZ, et al. Molecular characterization of human immunodeficiency virus type 1 and hepatitis C virus in paid blood donors and injection drug users in China[J]. J Virol, 2004, 78(24): 13591-13599. DOI:10.1128/JVI.78.24.13591-13599.2004 |
| [11] |
Xia XS, Lu L, Tee KK, et al. The Unique HCV Genotype distribution and the discovery of a novel subtype 6u among IDUs co-infected with HIV-1 in Yunnan, China[J]. J Med Virol, 2008, 80(7): 1142-1152. DOI:10.1002/jmv.21204 |
| [12] |
Agdamag DM, Kageyama S, Alesna ET, et al. Rapid spread of hepatitis C virus among injecting-drug users in the Philippines:implications for HIV epidemics[J]. J Med Virol, 2010, 77(2): 221-226. DOI:10.1002/jmv.20439 |
| [13] |
Shinji T, Kyaw YY, Gokan K, et al. Analysis of HCV genotypes from blood donors shows three new HCV type 6 subgroups exist in Myanmar[J]. Acta Med Okayama, 2004, 58(3): 135-142. DOI:10.18926/AMO/32110 |
| [14] |
Sacks-Davis R, Daraganova G, Aitken C, et al. Hepatitis C virus phylogenetic clustering is associated with the social-injecting network in a cohort of people who inject drugs[J]. PLoS One, 2012, 7(10): e47335. DOI:10.1371/journal.pone.0047335 |
| [15] |
古海尔·肉孜, 周素娟, 达吾提江·麦麦提, 等.新疆部分地区人类免疫缺陷病毒/丙型肝炎病毒合并感染者的丙型肝炎病毒5'端非编码区基因变异分析[J].中华传染病杂志, 2016, 34(3): 151-155. DOI: 10.3760/cma.j.issn.1000-6680.2016.03.004. Rouzi G, Zhou SJ, Maimaiti D, et al. Analysis of genetic variations of hepatitis C virus 5'untranslated regions gene in human immunodeficiency; virus-1/hepatitis C virus co-infected patients in Xinjiang[J]. Chin J Infect Dis, 2016, 34(3): 151-155. DOI: 10.3760/cma.j.issn.1000-6680.2016.03.004. |