 2018, Vol. 39
2018, Vol. 39文章信息
- 万永虎, 庄丽, 郑勤妮, 任丽娟, 付琳, 蒋维佳, 唐光鹏, 张德著, 李世军.
- Wan Yonghu, Zhuang Li, Zheng Qinni, Ren Lijuan, Fu Lin, Jiang Weijia, Tang Guangpeng, Zhang Dezhu, Li Shijun.
- 贵州省2014-2017年H7N9禽流感病毒HA和NA基因特征分析
- Genetic characteristics of hemagglutinin and neuraminidase of avian influenza A (H7N9) virus in Guizhou province, 2014-2017
- 中华流行病学杂志, 2018, 39(11): 1465-1471
- Chinese Journal of Epidemiology, 2018, 39(11): 1465-1471
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2018.11.009
-
文章历史
收稿日期: 2018-07-10




血凝素(Hemagglutinin,HA)和神经氨酸酶(Neuraminidase,NA)是流感病毒2个主要的包膜蛋白抗原,是甲型流感病毒亚型分类的主要依据[1-2],H7N9禽流感病毒是一个新型重组病毒,它一般仅在禽类之间流行,不易感染人类,但自2013年春季出现人感染H7N9禽流感病例之后,目前已发生了5波疫情[3],截止2017年10月26日,WHO共报告了1 564例人感染H7N9禽流感病例,其病死率约为40%。频繁的禽类迁徙和H7N9病毒在禽类中无症状传播,环境和自然选择压力及病毒本身因素的影响,可能导致病毒持续不断变异进化,这给禽类养殖业和人类健康构成了严重的威胁。2014-2017年贵州省共发生人感染H7N9禽流感病例20例,仅2017年就发生17例,死亡8例(本研究获得18例)。为了解贵州省H7N9禽流感病毒的病毒特征,本研究对2014-2017年贵州省H7N9禽流感病毒的HA和NA基因的特征和遗传进化进行了分析。
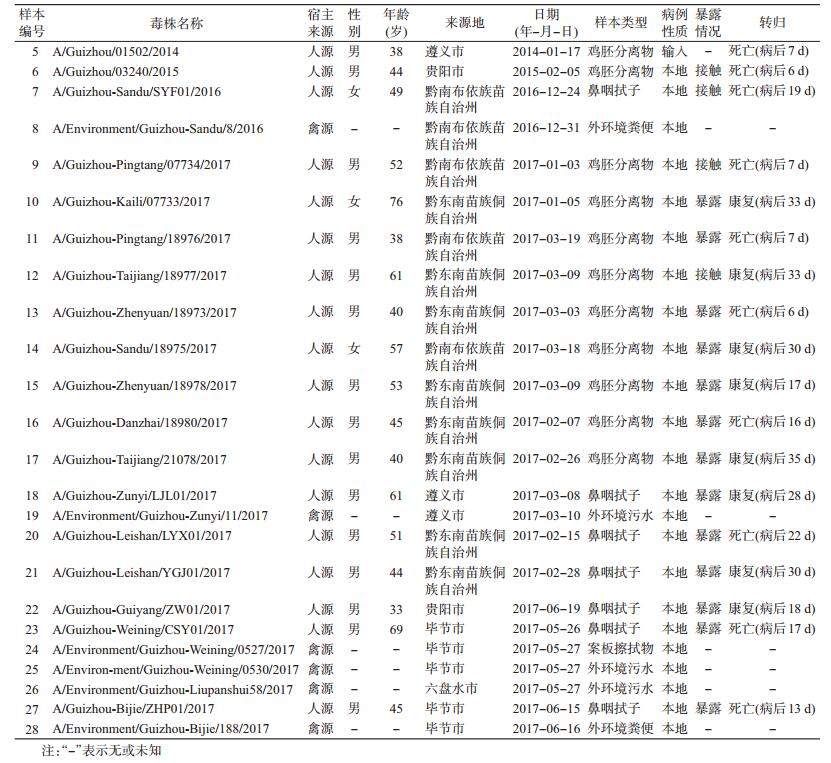
材料与方法1.材料来源:样本来自于2014-2017年贵州省18例人感染H7N9禽流感病例和6份活禽市场环境样本(表 1),本研究中其他人源、禽源和环境H7N9禽流感病毒参比序列来自于National Center of Biotechnology Information(NCBI)或the Global Initiative on Sharing Avian Influenza Data(GISAID)数据库,其中疫苗株参比序列编号和毒株名称分别为A/Shanghai/2/2013、A/Anhui/1/2013、A/Hunan/02650/2016、A/Guangdong/17SF003/2016。
2. HA和NA基因的扩增:采用西安天隆科技有限公司病毒DNA/RNA提取试剂盒提取鼻咽拭子和外环境样本总RNA,并用宝生物工程(大连)有限公司Prime Script One Step RT-PCR Kit Ver.2扩增HA和NA基因,产物经电泳分析确定后,送生工生物工程(上海)股份有限公司测序。
3.序列分析:采用Lasergene 7.1软件包中的EditSeq和SeqMan软件对测序结果进行编辑和拼接,采用MegAlign进行核苷酸同源性分析,然后采用MEGA 6.0软件对核苷酸推导出的氨基酸序列进行位点变异比对和对核苷酸进行进化树的构建,进化树采用Neighbor-Joining法,Bootstrap重复值为1 000。
结果1.一般情况:2014-2017年贵州省共发生20例人感染H7N9病例,2014-2016年各1例,2017年17例,共死亡11例,病死率为55.0%;该病主要发生在冬春季,集中分布在每年12月至次年3月,2017年5-6月发生了3例;主要分布在黔东南苗族侗族自治州(8例)、黔南布依族苗族自治州(5例)和毕节市(2例);发病年龄在33~76(M=47)岁,>60岁病例4例(20.0%),男女性别比为17 : 3;19例病例有与活禽或活禽市场暴露史;病例均为散发病例。
2. HA和NA基因同源性分析:与WHO推荐的不同时期4株疫苗株进行序列比对,2014-2015年贵州省的2株H7N9毒株与疫苗株A/Shanghai/2/2013和A/Anhui/1/2013的HA和NA基因的核苷酸同源性均较高,分别为98.8%~99.2%和99.2%;2016-2017年有16株毒株与疫苗株A/Hunan/02650/2016的HA和NA基因的核苷酸同源性最高,分别为98.2%~99.3%和97.6%~98.8%,2017年其余的6株与疫苗株A/Guangdong/17SF003/2016的HA和NA基因的核苷酸同源性最高,分别为99.1%~99.4%和98.9%~99.3%。
3. HA和NA基因遗传进化分析:贵州省24株毒株均属于长江三角洲分支,HA和NA基因进化树除2014-2015年的2株毒株分别成为一个小分支,且与疫苗株A/Shanghai/2/2013和A/Anhui/1/2013遗传距离较近外,其余22株毒株HA基因进化树聚集在3个次分支H7-1、H7-2和H7-3,NA基因进化树也聚集在3个次分支N9-1、N9-2和N9-3,其中HA基因进化树H7-1与H7-2分支和NA基因进化树N9-1与N9-2分支毒株均与疫苗株A/Hunan/02650/2016遗传距离较近,H7-1和N9-2分支毒株分别与此疫苗株处于同一次分支,而H7-3分支和N9-3分支毒株均与疫苗株A/Guangdong/17SF003/2016遗传距离较近,与其他分支毒株遗传距离较远(图 1,2)。

|
| 注:●疫苗株;▲贵州省人感染的H7N9毒株;△贵州省环境中的H7N9毒株 图 1 H7N9禽流感病毒HA基因遗传进化树 |

|
| 注:●疫苗株;▲贵州省人感染的H7N9毒株;△贵州省环境中的H7N9毒株 图 2 H7N9禽流感病毒NA基因遗传进化树 |
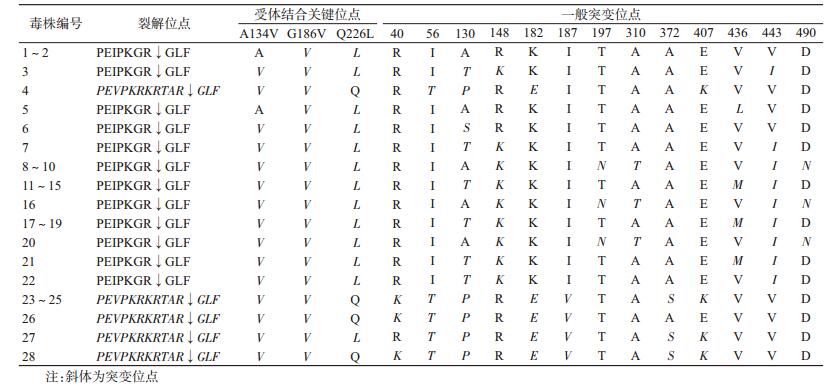
4. HA和NA蛋白分子特征分析:贵州省编号为23~28的6株毒株裂解位点发生插入突变为PEVPKRKRTAR↓GLF,含4个(KRKR)连续的碱性氨基酸,具备高致病性禽流感病毒的分子特征,其余18株毒株均为PEIPKGR↓GLF,仅含1个R连续的碱性氨基酸,为低致病性禽流感病毒分子特征。受体结合关键位点编号为6~28的23株毒株均发生A134V的突变,所有毒株均发生了G186V的突变,除编号为23~26和28的毒株未发生Q226L的突变以外,其余均发生Q226L的突变。此外,多个位点发生新的突变,包括R40K、I56T、A130T/P/S、R148K、K182E、I187V、T197N、A310T、A372S、E407K、V436L/M、V443I和D490N。所有毒株均含有5个糖基化位点为NGT、NAT、NDT、NWT和NNT,未发生数目和位点的突变,编号23~28的毒株后两个糖基化位点NWT和NNT发生了4个位置的后移(表 2)。
NA蛋白氨基酸位点所有毒株均存在QISNT的缺失;编号为16的毒株R294K耐药位点发生突变,其余耐药位点较稳定;此外,多个位点也发生新的突变,其中编号为23~28的毒株共同发生的突变有A21T、L29I、P39S、E78G、R131K、V207I、A359D和R432K,编号7~8和22毒株共同发生的突变有M77I和I305V,编号为9~10、16和20的毒株共同发生的突变有D126N、V238I、I335M、A370T和L399S,编号为11~15、17~19和21的毒株共同发生的突变有T183A和V214I。NA蛋白共含有7个糖基化位点,第一个糖基化位点编号为11~15、17~19和21的毒株发生突变NCS42NCT,其余较稳定,分别为52NTS、63NET、66NIT、87NLT、147NGT和202NAS(表 3)。

同源性分析显示,2014-2017年贵州省发现的H7N9禽流感病毒与不同的疫苗株相似度不同。H7N9禽流感暴发中心分为长江三角洲和珠江三角洲两处[4],贵州省24株毒株均属于长三角谱系,但却处于不同的次分支,2016-2017年毒株主要聚集在3个分支,疫苗株A/Hunan/02650/2016的HA基因与H7-1分支毒株遗传距离较近,NA基因却与N9-2分支毒株遗传距离较近,推测贵州省H7N9禽流感病毒HA和NA基因可能存在重配现象;而2017年贵州省毕节市和六盘水市的6株毒株与疫苗株A/Guangdong/17SF003/2016遗传距离较近,处于一个新的分支且与其他分支遗传距离较远;提示疫苗株对我国H7N9流行株的匹配性存在一定差距。
HA的关键氨基酸位点包括裂解位点、受体结合位点和糖基化位点,对宿主的致病性、特异性、抗宿主的免疫力和病毒的毒力都发挥着重要的作用[5-6]。蛋白分子特征分析显示,2017年贵州省西部2例人感染病例毒株裂解位点发生突变含4个(KRKR)连续的碱性氨基酸,突变为PEVPKRKRTAR↓GLF,属于高致病性禽流感病毒,由于此突变的H7N9毒株具备高致病性的分子特征,且在鸡群中具有高度的致病性和有效的传播性,对哺乳动物表现出快速的适应性[7-8],在鼠类和雪貂中也表现出较高的致病性、致死性和有效的传播性以及对神经氨酸酶抑制剂作用的有限性[9],感染的2例病例在2017年5-6月非流行季节,2例病例分别病后13 d和17 d均死亡,推测可能与毒株的高致病性有关。若此突变占据主要优势,将有可能形成潜在的流感大流行。贵州省H7N9病毒受体结合关键位点主要的突变是134、186和226位点;据文献报道,A134V的突变有利于病毒的复制与包装,而G186V和Q226L的突变有利于病毒与人类上呼吸道α-2,6唾液酸受体的结合[10],贵州省23株H7N9毒株发生A134V的突变,24株均发生了G186V的突变,19株含1株高致病性毒株发生Q226L的突变,提示贵州省H7N9禽流感病毒株与人样受体结合能力增强,且高致病性毒株正在发生有利于向人类传播的进化。
贵州省24株H7N9毒株NA蛋白均存在“QISNT”的缺失,这使得H7N9病毒适应了陆生家禽且毒力增强[11]。贵州省23株H7N9毒株其耐药位点较稳定,未发生突变,但毒株A/Guizhou-Danzhai/18980/2017在R294K耐药位点发生突变,提示该毒株对奥司他韦和扎那米韦神经氨酸酶抑制剂药物产生了耐药[12],耐药突变位点的产生可能是药物压力选择下的结果,仍需进一步的确认,提示抗病毒药物也应该合理使用。NA蛋白其他位点也发生了一些新的突变,具体的功能作用有待进一步的研究。贵州省9株H7N9毒株糖基化位点发生NCS42NCT突变,可能影响病毒的毒力和宿主特异性[13]。此外,HA和NA蛋白上还发生了一些新的突变,亟待我们明确这些突变对病毒产生的影响。
高致病性H7N9禽流感病毒株和耐药株的出现,提示应加强对病毒基因的突变监测研究,密切关注其在人群中的演化,早期发现并加以控制是阻断禽流感流行的关键。
利益冲突 无
| [1] |
Li Q, Sun XM, Li ZX, et al. Structural and functional characterization of neuraminidase-like molecule N10 derived from bat influenza A virus[J]. Proc Natl Acad Sci USA, 2012, 109(46): 18897-18902. DOI:10.1073/pnas.1211037109 |
| [2] |
Tong SX, Zhu XY, Li Y, et al. New world bats harbor diverse influenza A viruses[J]. PLoS Pathog, 2013, 9(10): 1-12. DOI:10.1371/journal.ppat.1003657 |
| [3] |
Su S, Gu M, Liu D, et al. Epidemiology, evolution, and pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China[J]. Trends Microbiol, 2017, 25(9): 713-728. DOI:10.1016/j.tim.2017.06.008 |
| [4] |
Wang DY, Yang L, Zhu WF, et al. Two outbreak sources of influenza A (H7N9) viruses have been established in China[J]. J Virol, 2016, 90(12): 5561-5573. DOI:10.1128/JVI.03173-15 |
| [5] |
Matrosovich MN, Gambaryan AS, Teneberg S, et al. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor-binding site[J]. Virology, 1997, 233(1): 224-234. DOI:10.1006/viro.1997.8580 |
| [6] |
余慧燕, 许可, 邓斐, 等. 2016-2017年江苏省人感染H7N9禽流感病毒HA和NA基因分子进化分析[J]. 病毒学报, 2017, 33(5): 685-690. Yu HY, Xu K, Deng F, et al. Molecular characteristics of the HA and NA genes of the human influenza A (H7N9) virus in Jiangsu province, China, 2016-2017[J]. Chin J Virol, 2017, 33(5): 685-690. DOI:10.13242/j.cnki.bingduxuebao.003219 |
| [7] |
Qi W, Jia W, Liu D, et al. Emergence and adaptation of a novel highly pathogenic H7N9 influenza virus in birds and humans from a 2013 human-infecting low-pathogenic ancestor[J]. J Virol, 2018, 92(2): e00921-17. DOI:10.1128/JVI.00921-17 |
| [8] |
Iuliano AD, Jang Y, Jones J, et al. Increase in human infections with avian influenza A(H7N9) virus during the fifth epidemic-China, October 2016-February 2017[J]. MMWR Morb Mortal Wkly Rep, 2017, 66(9): 254-255. DOI:10.15585/mmwr.mm6609e2 |
| [9] |
Shi JZ, Deng GH, Kong HH, et al. H7N9 virulent mutants detected in chickens in China pose an increased threat to humans[J]. Cell Res, 2017, 27(12): 1409-1421. DOI:10.1038/cr.2017.129 |
| [10] |
Xiong XL, Martin SR, Haire LF, et al. Receptor binding by an H7N9 influenza virus from humans[J]. Nature, 2013, 499(7459): 496-499. DOI:10.1038/nature12372 |
| [11] |
Yang H, Carney PJ, Chang JC, et al. Structural analysis of the hemagglutinin from the recent 2013 H7N9 influenza virus[J]. J Virol, 2013, 87(22): 12433-12446. DOI:10.1128/JVI.01854-13 |
| [12] |
Chen JD, Zhang JP, Zhu WJ, et al. First genome report and analysis of chicken H7N9 influenza viruses with poly-basic amino acids insertion in the hemagglutinin cleavage site[J]. Scientific Reports, 2017, 7: 9972. DOI:10.1038/s41598-017-10605-6 |
| [13] |
Chen WT, Zhong YG, Qin YN, et al. The evolutionary pattern of glycosylation sites in influenza virus (H5N1) hemagglutinin and neuraminidase[J]. PLoS One, 2012, 7(11): e49224. DOI:10.1371/journal.pone.0049224 |