 2018, Vol. 39
2018, Vol. 39文章信息
- 李春晓, 高莹, 高文静, 余灿清, 吕筠, 吕若然, 段佳丽, 孙颖, 郭向晖, 王胜锋, 周斌, 王观, 曹卫华, 李立明.
- Li Chunxiao, Gao Ying, Gao Wenjing, Yu Canqing, Lyu Jun, Lyu Ruoran, Duan Jiali, Sun Ying, Guo Xianghui, Wang Shengfeng, Zhou Bin, Wang Guan, Cao Weihua, Li Liming.
- 7~16岁儿童青少年肥胖与DNA甲基化相关性的双生子研究
- Association between obesity and DNA methylation among the 7-16 year-old twins
- 中华流行病学杂志, 2018, 39(4): 443-448
- Chinese Journal of Epidemiology, 2018, 39(4): 443-448
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2018.04.011
-
文章历史
收稿日期: 2017-09-12




2. 100013 北京市疾病预防控制中心;
3. 100021 北京市朝阳区疾病预防控制中心
2. Beijing Center for Disease Control and Prevention, Beijing 100013, China;
3. Chaoyang District Center for Disease Control and Prevention, Beijing 100021, China
近年来,我国7~18岁儿童青少年超重肥胖检出率呈上升趋势[1-3]。DNA甲基化是研究较多的表观遗传现象,是指在受到上游遗传调控或下游环境因素影响时,胞嘧啶C的5’碳位上共价结合一个甲基基团,形成5-甲基胞嘧啶,导致DNA序列未发生变化但基因功能发生了可遗传的改变,可以同时反映遗传和环境的作用[4]。研究肥胖相关DNA甲基化位点,可为探索肥胖病因机制开拓思路,已成为近年来的研究热点[5]。一般人群的甲基化研究难以控制遗传因素,研究所需样本量大,进行大规模的全基因组甲基化芯片检测较昂贵。表型不一致的同卵双生子研究与全基因组DNA甲基化研究相结合,既可以利用配对双生子天然匹配基因背景和共同环境因素(如宫内环境),减少研究所需样本量[6];又可以通过先进的高通量甲基化检测芯片(美国Illumina公司的850 K甲基化EPIC芯片),在小样本、节约资金的基础上,最大限度地覆盖全基因组甲基化位点,从而在分析基因-环境交互作用背后的表观遗传学机制中具有特殊优势。同时,儿童青少年与成年人相比,可以更好地避免吸烟等复杂环境因素对甲基化水平的干扰,更易探索影响早期肥胖发生的因素及表观遗传学机制,对于控制肥胖及相关疾病的发生具有重要意义[5]。目前,儿童青少年肥胖的全基因组甲基化研究开展较少;由于样本的难获得性,肥胖不一致双生子研究更为少见。已有研究应用上一代450 K芯片发现了许多位点在肥胖组与非肥胖组间存在差异,但多重校正后无满足显著性水平要求的阳性位点[7-10]。是否存在与中国儿童青少年肥胖相关的DNA甲基化位点有待证实。因此,本研究利用2016年在北京市收集的7~16岁肥胖不一致同卵双生子,采用850 K芯片,在全基因组水平上探索与中国儿童青少年肥胖相关的DNA甲基化位点。
对象与方法1.研究对象:基于2016年在北京市朝阳区、延庆区及房山区招募的90对6~17周岁双生子,从中挑选肥胖不一致同卵双生子(MZ)。所有流程通过北京大学医学部伦理委员会审查(审查编号:IRB00001052-15029)。双生子成员均为北京市常住居民,均能提供基本信息、配合体检,并成功收集其外周血样本,双生子家长均签署纸质知情同意书。
为利用疾病不一致MZ天然匹配的优势,发现肥胖相关的DNA甲基化位点,本研究结合以往研究中成年双生子肥胖不一致标准制定方法[11-12],并参考中国儿童BMI及腰围(WC)分类标准[13-14],找出肥胖不一致(如正常-超重、正常-肥胖、超重-肥胖,或肥胖-肥胖、超重-超重且体格指标有差异)的双生子对。其中BMI和WC不一致条件两者满足其一即可,都满足时优先按BMI分组。经过上述标准筛选后,最终本研究共纳入肥胖不一致双生子23对。
2.研究方法:肥胖不一致MZ可视为1:1匹配病例对照;DNA甲基化水平为连续变量,样本量可按配对t检验要求计算。根据现有文献,按甲基化水平在肥胖组和非肥胖组的平均差值为1%,s为1%~4%,α=0.05,power=80%,采用PASS 11.0软件,计算样本量最少为10对,最大为128对[15]。本研究纳入7~16岁肥胖不一致MZ 23对。
问卷调查信息由调查员面对面访问获得,其中一般人口学信息(如性别、出生日期等)由双生子父母提供。身高、体重(百利达MC-780人体成分测量仪)和WC等体格指标由经过统一培训的调查员采用统一工具测量获得。上述测量指标读数分别以cm和kg为单位,精确到0.1,进一步计算BMI=体重(kg)/身高(m)2。双生子血样由专业护士采集,于早晨收集空腹12 h后的外周血2 ml,于-80 ℃超低温冰箱中保存以备检测。全基因组DNA甲基化检测采用Illumina甲基化EPIC芯片(Illumina Human Methylation EPIC BeadChip)。DNA提取及甲基化检测由北京怡美通德科技发展有限公司完成。卵型鉴定采用甲基化芯片法测定[16]。
质量控制:本研究参考中国双生子登记系统(Chinese National Twin Registry,CNTR)[17],制定出标准化的调查工作手册,并经专家提出意见进行修改。对调查人员统一培训,严格质量控制。获得问卷后采用双人双录入,之后进行数据复核,保证数据真实、统一、准确。DNA甲基化检测质量控制根据现有标准在所有样本中进行[18-20],利用R 3.3.1软件minfi程序包进行甲基化数据提取与转换得到甲基化水平β值,剔除不合格样本和甲基化位点;再利用R 3.3.1软件wateRmelon程序包进行β值标准化,控制芯片本身的探针测量偏倚和背景噪声偏倚等,形成待分析的甲基化数据。最终用于本分析的合格样本46个(23对MZ),CpG位点817 471个。
3.统计学分析:采用经验贝叶斯配对调整t检验[21],利用R 3.3.1软件中limma程序包ebayes函数[22],分析DNA甲基化水平在肥胖指标较高组和较低组之间差异是否有统计学意义,探索与肥胖相关的甲基化差异CpG位点(differentially methylated CpG sites,DMCs)。采用经验贝叶斯配对调整Levene检验[21],利用R 3.3.1软件中missMethyl程序包varFit函数[23],分析肥胖组和非肥胖组组间的甲基化变异水平(方差)差异是否有统计学意义,探索肥胖相关的甲基化变异差异CpG位点(differentially variable CpG sites,DVCs)。采用代理变量分析方法(surrogate variable analysis,SVA),利用R 3.3.1软件中sva程序包,调整甲基化检测实验批次等潜在混杂[24-25]。本研究所有分析均在全基因组水平上进行,因分析位点较多,需要进行多重检验校正,显著性水平为错误发现阳性位点率(false discovery rate,FDR)<0.05,FDR按照Benjamini & Hochberg法[26]。敏感性分析:利用R 3.3.1软件ChAMP程序包控制白细胞组成成分估计值对结果的影响后[27-29],再进行上述分析,对比分析前后结果差异是否有统计学意义。
结果1.一般特征:根据本研究肥胖不一致标准,共收集23对肥胖不一致MZ,其中朝阳区9对,延庆区3对,房山区11对。其中,男性为12对,女性为11对,年龄为7~16(11.2±2.4)岁。研究对象的BMI、WC的x±s分别为(22.3±4.2)kg/m2、(75.4±10.7)cm。见表 1。纳入的肥胖不一致双生子:“肥胖-正常”双生子1对,“超重-正常”双生子8对,“肥胖-超重”双生子4对,均肥胖或均正常但肥胖指标存在差异的分别为9对和1对。BMI、WC在肥胖指标较高组和较低组间的平均差值为1.49 kg/m2、4.60 cm,差异均有统计学意义(P<0.05)。全基因组的CpG位点在基因结构分布中主要位于基因主体(body,37.1%),3’非编码区(3’-UTR,2.5%)分布最少;此外,分别有18.8%和18.0%的位点位于CpG岛(island)和CpG岸(shore),这些位置CpG位点相对比较密集,见图 1。


|
| 注:TSS1500:转录起始点1 500 bp范围内;TSS200:转录起始点200 bp范围内;5’UTR:5’非编码区;1st exon:第一外显子区;Body:基因主体;3’UTR:3’非编码区;CpG岛:人类基因组中大小为100~1 000 bp且富含CpG位点的区域,主要位于启动子或第一外显子区域;CpG岸:距离CpG岛2 000 bp的区域;CpG架:CpG岸外侧离岸2 000 bp的距离 图 1 全基因组817 471个CpG位点的基因组分布 |
2.肥胖不一致MZ对内全基因组DNA甲基化差异(DMCs)分析:肥胖组与非肥胖组的平均β值在绝大部分位点均存在差异,差异无统计学意义(FDR>0.05),差异最明显位点的原始P值接近1×10-6。见图 2。利用经验贝叶斯配对调整t检验,所有位点校正后的FDR均>0.05。P值最小的前10个位点及位置关系等信息见表 2。显著性水平前10位的位点中,7个为850 K芯片新增位点,此前研究中均未覆盖,多数CpG位点位于基因主体和基因的启动子区域。其中,P值最小位点为位于12号染色体上的位点cg05684382(P=1.26E-06)。cg05684382处于高甲基化水平(β>0.75),肥胖组的甲基化水平低于非肥胖组,差异无统计学意义。

|
| 图 2 肥胖不一致同卵双生子组间甲基化差异分布火山图 |
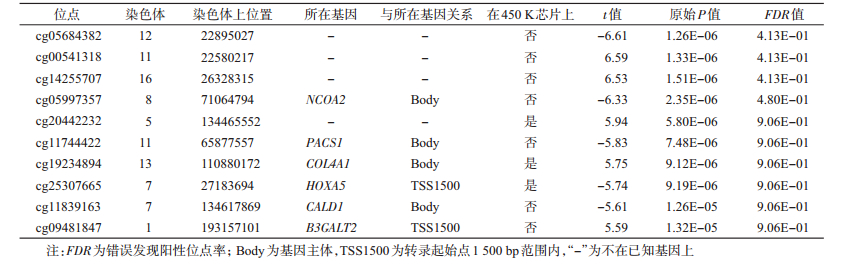
3.肥胖不一致MZ组间全基因组DNA甲基化变异差异(DVCs)分析:Levene检验未发现肥胖相关的甲基化变异性阳性位点,所有位点校正后的FDR均>0.05。P值最小的前10个位点大部分为850 K芯片新增位点,主要位于基因主体区域,其中P值最小位点为位于16号染色体CMIP基因上的位点cg26188191(P=6.44E-06)。见表 3。
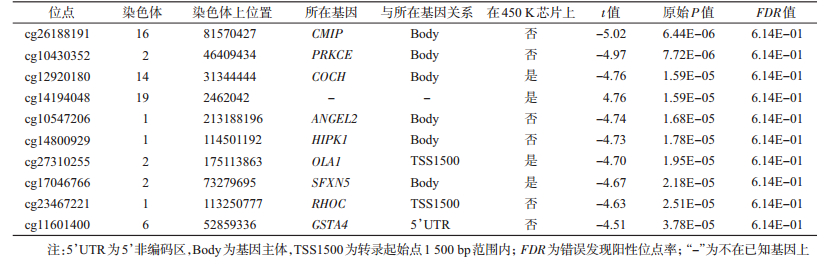
4.敏感性分析:血液组织的甲基化水平会受到血细胞成分组成的影响,因此利用Houseman的模型调整细胞组成进行敏感性分析,DMCs和DVCs均未显示有统计学意义的差异位点。在敏感性分析中P值较小的前10个位点与原始分析结果前10的位点无重合。此外研究还分别对DMCs和DVCs敏感性分析前后结果进行比较,发现P值最小的DMC位点cg05684382和DVC位点cg26188191在敏感性分析前后关联方向及趋势一致(结果未展示)。
讨论本研究针对7~16岁中国儿童青少年,利用肥胖不一致MZ这一天然匹配人群,控制遗传因素和共同环境因素的影响后,在全基因组水平上,探索肥胖相关DNA甲基化位点,是研究儿童青少年肥胖表观遗传学病因机制的有益尝试。目前对于儿童青少年肥胖的全基因组甲基化关联研究多来自西方国家的一般人群,由于人群的特殊性和难获得性,罕有肥胖不一致双生子研究设计。
本研究利用23对7~16岁肥胖不一致MZ为研究对象,采用国际上先进的850 K芯片,经过严格质量控制,在全基因组范围内的817 471个CpG位点中,探索肥胖与甲基化的相关性。经过多重校正未发现肥胖相关阳性位点,但研究提示某些位点可能与肥胖发生存在关联,其中DMC位点cg05684382和DVC位点cg26188191在肥胖不一致MZ中P值相对较小,且二者均为850 K芯片中新增的位点,以往全基因组甲基化研究中所用芯片皆为Illumina 27 K或450 K芯片,尚未见与这两个位点相关研究报道,提示后续研究可以重点关注。
本研究未发现与肥胖相关的DNA甲基化位点,与国际上大部分儿童肥胖全基因组甲基化关联研究的结果一致[7-10]。此前有研究利用候选基因策略,结果显示,部分位点与儿童青少年肥胖相关,但这些位点均未在全基因组甲基化关联研究中证实。目前尚不能确定儿童青少年肥胖与DNA甲基化是否相关,可能与以下因素有关。首先是遗传因素,研究显示DNA结构能够影响甲基化水平[30-32]。本研究利用MZ人群,排除大部分遗传因素影响后,未发现与肥胖存在关联的位点。因此以往候选基因研究中发现的儿童青少年肥胖相关的阳性位点,可能是上游遗传因素影响肥胖发生的一个中间环节,而非由环境因素作用或环境-遗传交互作用影响甲基化从而导致与肥胖的相关。其次,虽然双生子研究可以匹配掉很多混杂因素,也可能存在共同环境因素(如生命早期共同环境因素)介导了肥胖与DNA甲基化之间的相关,双生子匹配共同环境因素以后会使得二者关联结果为阴性[33],但是这种假设有可能成立的前提是在样本量绝对充足但仍未发现阳性位点时。第三,DNA甲基化存在组织特异性,肥胖表型不同的组间甲基化差异可能在脂肪组织中更能凸显。本研究采用外周血样本,虽然既往基于450 K芯片的成年人肥胖全基因组甲基化关联研究发现血细胞与在脂肪组织中发现的关联一致[34],但在儿童青少年中,尤其是805 K芯片最新覆盖位点的组织间差异尚不明确,因此需要考虑组织特异性对结果的影响。
本研究存在局限性。主要体现在样本量可能不足。本研究利用儿童青少年肥胖不一致双生子进行肥胖与DNA甲基化关联研究,尽管样本量理论上可以满足统计学效力的要求,但本研究中23对肥胖不一致双生子并非严格按照“肥胖-正常”筛选,可能会降低统计学效力;同时儿童青少年不同年龄段肥胖判定标准不同,因此无法像成年人研究一样简单设定BMI差距3 kg/cm2为肥胖不一致标准,增加了筛选合格样本的难度。但本研究可为后续此类研究计算样本量做参考。此外本研究仅采用外周血样本进行甲基化检测,并且无白细胞细胞分类的检验数据进行调整。但与当前国外研究保持一致,从方法学上按照国外同类研究常用的Houseman模型进行敏感性分析调整细胞成分后也未发现关联结果出现改变。
综上所述,本研究利用肥胖不一致双生子,在全基因组范围内针对儿童青少年进行了肥胖与DNA甲基化相关性的探索和尝试,虽然基于现有的研究样本,尚未发现满足统计学意义要求的阳性位点,但仍提示可能存在以往研究未发现的与肥胖存在关联的DNA甲基化位点,后续研究可以继续关注。本研究结果有待进一步验证,也可为后续此类利用肥胖不一致双生子的研究提供参考。
志谢: 感谢北京市房山区中小学卫生保健所林宁翔、延庆区中小学卫生保健所孟庆军和延庆区CDC王绍华等参与本次现场调查利益冲突: 无
| [1] | Gillman MW, Ludwig DS. How early should obesity prevention start?[J]. N Engl J Med, 2013, 369(23): 2173–2175. DOI:10.1056/NEJMp1310577 |
| [2] | Gortmaker SL, Swinburn BA, Levy D, et al. Changing the future of obesity:science, policy, and action[J]. Lancet, 2011, 378(9793): 838–847. DOI:10.1016/S0140-6736(11)60815-5 |
| [3] |
马军, 蔡赐河, 王海俊, 等. 1985-2010年中国学生超重与肥胖流行趋势[J]. 中华预防医学杂志, 2012, 46(9): 776–780.
Ma J, Cai CH, Wang HJ, et al. The trend analysis of overweight and obesity in Chinese students during 1985-2010[J]. Chin J Prev Med, 2012, 46(9): 776–780. DOI:10.3760/cma.j.issn.0253-9624.2012.09.002 |
| [4] | Bird A. Perceptions of epigenetics[J]. Nature, 2007, 447(7143): 396–398. DOI:10.1038/nature05913 |
| [5] |
高莹, 高文静, 曹卫华. 儿童青少年肥胖与DNA甲基化关联研究进展[J]. 中华流行病学杂志, 2016, 37(8): 1169–1174.
Gao Y, Gao WJ, Cao WH. Current status and progress in studies on the association between obesity and DNA methylation among children and adolescents[J]. Chin J Epidemiol, 2016, 37(8): 1169–1174. DOI:10.3760/cma.j.issn.0254-6450.2016.08.023 |
| [6] |
王碧琦, 周斌, 高文静, 等. 双生子在表观遗传学研究中的价值[J]. 中华流行病学杂志, 2015, 36(4): 402–404.
Wang BQ, Zhou B, Gao WJ, et al. The value of twin study in epigenetic research[J]. Chin J Epidemiol, 2015, 36(4): 402–404. DOI:10.3760/cma.j.issn.0254-6450.2015.04.023 |
| [7] | Wang XL, Zhu HD, Snieder H, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes[J]. BMC Med, 2010, 8: 87. DOI:10.1186/1741-7015-8-87 |
| [8] | Ding X, Zheng DY, Fan CN, et al. Genome-wide screen of DNA methylation identifies novel markers in childhood obesity[J]. Gene, 2015, 566(1): 74–83. DOI:10.1016/j.gene.2015.04.032 |
| [9] | Huang RC, Garratt ES, Pan H, et al. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood[J]. Epigenetics, 2015, 10(11): 995–1005. DOI:10.1080/15592294.2015.1080411 |
| [10] | Rounge TB, Page CM, Lepisto M, et al. Genome-wide DNA methylation in saliva and body size of adolescent girls[J]. Epigenomics, 2016, 8(11): 1495–1505. DOI:10.2217/epi-2016-0045 |
| [11] | Bondia-Pons I, Maukonen J, Mattila I, et al. Metabolome and fecal microbiota in monozygotic twin pairs discordant for weight:a Big Mac challenge[J]. FASEB J, 2014, 28(9): 4169–4179. DOI:10.1096/fj.14-250167 |
| [12] | Marniemi J, Kronholm E, Aunola S, et al. Visceral fat and psychosocial stress in identical twins discordant for obesity[J]. J In Med, 2002, 251(1): 35–43. DOI:10.1046/j.1365-2796.2002.00921.x |
| [13] |
马冠生, 季成叶, 马军, 等. 中国7~18岁学龄儿童青少年腰围界值点研究[J]. 中华流行病学杂志, 2010, 31(6): 609–615.
Ma GS, Ji CY, Ma J, et al. Waist circumference reference values for screening cardiovascular risk factors in Chinese children and adolescents aged 7-18 years[J]. Chin J Epidemiol, 2010, 31(6): 609–615. DOI:10.3760/cma.j.issn.0254-6450.2010.06.003 |
| [14] |
中国肥胖问题工作组. 中国学龄儿童青少年超重、肥胖筛查体重指数值分类标准[J]. 中华流行病学杂志, 2004, 25(2): 97–102.
China's Working Group on Obesity. Body mass index reference norm for screening overweight and obesity in Chinese children and adolescents[J]. Chin J Epidemiol, 2004, 25(2): 97–102. DOI:10.3760/j.issn:0254-6450.2004.02.003 |
| [15] | Zhao J, Goldberg J, Vaccarino V. Promoter methylation of serotonin transporter gene is associated with obesity measures:a monozygotic twin study[J]. Int J Obesity, 2013, 37(1): 140–145. DOI:10.1038/ijo.2012.8 |
| [16] | Wang BQ, Gao WJ, Yu CQ, et al. Determination of zygosity in adult Chinese twins using the 450 K methylation array versus questionnaire data[J]. PLoS One, 2015, 10(4): e0123992. DOI:10.1371/journal.pone.0123992 |
| [17] |
高文静, 李立明. 中国双生子队列研究进展[J]. 中华流行病学杂志, 2017, 38(6): 828–831.
Gao WJ, Li LM. The Chinese national twin cohort:an update[J]. Chin J Epidemiol, 2017, 38(6): 828–831. DOI:10.3760/cma.j.issn.0254-6450.2017.06.027 |
| [18] | Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi:a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays[J]. Bioinformatics, 2014, 30(10): 1363–1369. DOI:10.1093/bioinformatics/btu049 |
| [19] | McCartney DL, Walker RM, Morris SW, et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium Methylation EPIC BeadChip[J]. Genomics Data, 2016, 9: 22–24. DOI:10.1016/j.gdata.2016.05.012 |
| [20] | Pidsley R, Wong CCY, Volta M, et al. A data-driven approach to preprocessing Illumina 450 K methylation array data[J]. BMC Genom, 2013, 14: 293. DOI:10.1186/1471-2164-14-293 |
| [21] | Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments[J]. Stat Appl Genet Mol Biol, 2004. DOI:10.2202/1544-6115.1027 |
| [22] | Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies[J]. Nucleic Acids Res, 2015, 43(7): e47. DOI:10.1093/nar/gkv007 |
| [23] | Phipson B, Oshlack A. DiffVar:a new method for detecting differential variability with application to methylation in cancer and aging[J]. Genome Biol, 2014, 15(9): 465. DOI:10.1186/s13059-014-0465-4 |
| [24] | Leek JT, Johnson WE, Parker HS, et al. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments[J]. Bioinformatics, 2012, 28(6): 882–883. DOI:10.1093/bioinformatics/bts034 |
| [25] | Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis[J]. PLoS Genet, 2007, 3(9): 1724–1735. DOI:10.1371/journal.pgen.0030161 |
| [26] | Benjamini Y, Hochberg Y. Controlling the false discovery rate-a practical and powerful approach to multiple testing[J]. J Royal Stat Soc Ser B-Methodol, 1995, 57(1): 289–300. |
| [27] | Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution[J]. BMC Bioinf, 2012, 13: 86. DOI:10.1186/1471-2105-13-86 |
| [28] | Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies[J]. Genome Biol, 2014, 15(2): R31. DOI:10.1186/gb-2014-15-2-r31 |
| [29] | Morris TJ, Butcher LM, Feber A, et al. ChAMP:450 K chip analysis methylation pipeline[J]. Bioinformatics, 2014, 30(3): 428–430. DOI:10.1093/bioinformatics/btt684 |
| [30] | Bell JT, Pai AA, Pickrell JK, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines[J]. Genome Biol, 2011, 12(6): 405. DOI:10.1186/gb-2011-12-1-r10 |
| [31] | Drong AW, Nicholson G, Hedman ÅK, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue[J]. PLoS One, 2013, 8(2): e55923. DOI:10.1371/journal.pone.0055923 |
| [32] | Kerkel K, Spadola A, Yuan E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation[J]. Nat Genet, 2008, 40(7): 904–908. DOI:10.1038/ng.174 |
| [33] | Tobi EW, Slieker RC, Stein AD, et al. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome[J]. Int J Epidemiol, 2015, 44(4): 1211–1123. DOI:10.1093/ije/dyv043 |
| [34] | Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index:a genome-wide analysis[J]. Lancet, 2014, 383(9933): 1990–1998. DOI:10.1016/s0140-6736(13)62674-4 |