 2018, Vol. 39
2018, Vol. 39文章信息
- 王梦莹, 刘冬静, 黄辉, 李文咏, 周仁, 朱洪平, 周治波, 吴涛.
- Wang Mengying, Liu Dongjing, Huang Hui, Li Wenyong, Zhou Ren, Zhu Hongping, Zhou Zhibo, Wu Tao.
- 非综合征型唇腭裂二代测序研究进展
- Progress in next-generation sequencing research of non-syndromic oral clefts
- 中华流行病学杂志, 2018, 39(3): 387-390
- Chinese Journal of Epidemiology, 2018, 39(3): 387-390
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2018.03.026
-
文章历史
收稿日期: 2017-07-23




2. 100081 北京大学口腔医院颌面外科
2. Department of Oral and Maxillofacial Surgery, School of Stomatology, Peking University, Beijing 100081, China
唇腭裂(oral clefts)是由于胚胎发育过程中颌腭组织发育不全或发育受阻而引起的一组颌面部先天性畸形[1]。唇腭裂可按解剖结构分为单纯腭裂(cleft palate,CP)、单纯唇裂(cleft lip,CL)和唇裂合并腭裂(cleft lip with cleft palate,CLP)[2]。CL和CLP由于流行病学特征以及胚胎发育起源上的相似性,常被归为一类,称为唇裂合并或不合并腭裂(cleft lip with or without cleft palate,CL/P)[3]。此外,唇腭裂还可分为综合征型与非综合征型两类,前者为单基因遗传病,而非综合征型唇腭裂(non-syndromic oral clefts,NSOC)患儿不伴有其他畸形或异常,是受多基因影响的复杂疾病[2]。NSOC更为常见,约70%的CL/P患者及50%的CP患者为非综合征型[4]。唇腭裂活产儿的患病率约为1/700[5],中国人群的唇腭裂患病率为1.49/1 000活产儿~1.56/1 000活产儿,NSOC的患病率为1.13/1 000活产儿~1.30/1 000活产儿,高于其他地区和种族人群[6]。作为一种病因复杂的疾病,虽然全基因组关联研究发现了NSOC的多个遗传致病位点,但目前发现的遗传位点只能解释NSOC一小部分遗传度,而二代测序研究为进一步探索NSOC遗传危险因素提供了新的方法和思路,本文将对NSOC二代测序研究进行综述。
一、NSOC遗传流行病学研究现状生物标志物检测方法的迅速发展使唇腭裂易感基因的定位成为可能。全基因组关联研究(genome-wide association study,GWAS)是复杂疾病的高效定位方法。随着GWAS在NSOC领域的开展,研究者发现了在候选基因研究年代从未观察到的强关联区域。GWAS不仅验证了一些与NSOC有关的候选基因,同时还发现了很多新位点如8q24、MAFB、VAX1等。基于GWAS开展的Meta分析则通过最大程度地整合现有证据,深入挖掘常见遗传变异的致病效应。GWAS Meta分析中发现和验证的危险等位基因[7-8],见表 1。
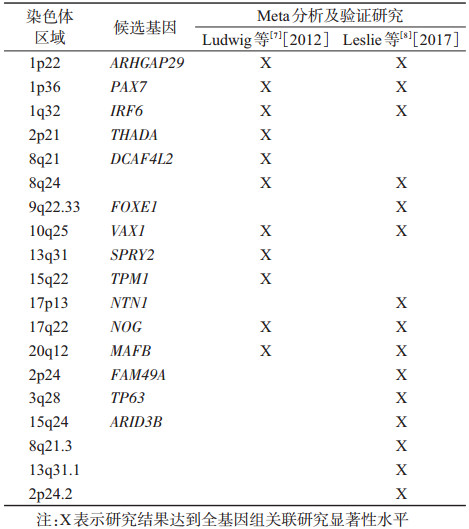
虽然GWAS研究发现了NSOC的多个遗传危险位点,但如其他复杂疾病一样,迄今发现的遗传变异仅能解释NSOC很少一部分的遗传度。GWAS理论基础是常见疾病可归因于人群中弱势等位基因频率(minor allele frequency,MAF)高于1%的常见遗传变异(common variants)[9-10],从设计原理上无法发现频率低但具有较大致病效应的罕见遗传变异以及新发突变。因此,搜寻罕见遗传变异、新发突变等具有较强致病效应的位点、揭示遗传危险因素的致病机制成为后GWAS时代NSOC病因研究的焦点。
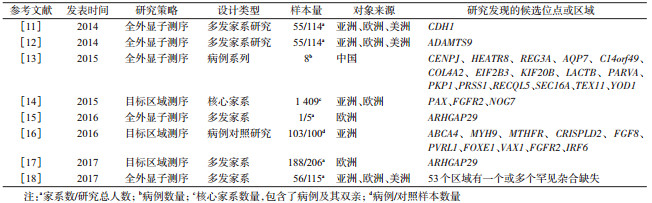
二、NSOC二代测序研究二代测序技术(next generation sequencing,NGS)可以获得大量罕见遗传变异,为解决上述问题提供了新思路和新方法,一些无法由GWAS发现的NSOC罕见遗传变异和新发突变,有望通过NGS方法搜寻发现。目前已有多种针对NSOC的二代测序研究策略,主要包括全基因组测序(whole genome sequencing,WGS)、全外显子测序(whole exome sequencing,WES)以及目标区域测序(target sequencing)。目前主要的NSOC二代测序研究结果[11-18],见表 2。
1. NSOC的WGS研究:WGS是指对人类整个基因组进行测序,具有更高的覆盖均匀性,可以更好地识别拷贝数变异(copy number variation,CNV),插入缺失(insertion and deletion,InDel)和基因融合等。此外,WGS还覆盖基因组的非编码区域,包括内含子、启动子、非编码区和调控元件等。因此,它可以更全面地揭示疾病的分子改变[19-20]。虽然由于成本较高、所需时间较长以及产生的数据容量较大等原因,WGS还没有得到广泛的应用,但WGS在基因组覆盖以及富集策略等方面仍然占有一定优势。Belkadi等[21]将6例患者的WGS和WES数据进行比较,结果发现WGS数据质量较好,并且突变的检出率更高。虽然目前尚没有针对NSOC的WGS,但大规模的WGS研究正在进行中,将用于NSOC等复杂性疾病的病因探索[22]。
2. NSOC WES研究:人类基因组共有30亿个碱基对(base pair,bp),其中约1%碱基对(3 000万bp)构成编码序列。对基因组编码区(外显子组)进行测序有助于NSOC等复杂疾病功能性致病基因位点的发现。随着相应统计分析方法的发展,WES已成为一种可靠的发现疾病致病位点的重要方法,并将迅速发展到对全基因组的测序研究,为最终解决复杂疾病的病因学提供思路。
WES技术适用于少量家系样本的研究,自美国华盛顿大学学者Ng等[23]在2009年首次利用WES研究分析单基因遗传病以来,WES已被应用于NSOC等多种复杂疾病遗传危险因素的探索。Bureau等[11]于2014年开展了以NSOC多发家系为样本的全外显子组测序研究,结果显示来自一个家庭中3个家系成员共同携带的CDH1基因中某一罕见单核苷酸变异(single nucleotide variation,SNV)达到了显著性水平,该变异很有可能是NSOC的致病遗传变异。CDH1基因在细胞黏附中发挥重要作用,对颅面形态形成和腭融合等生长发育至关重要[24]。研究者在4个不同人群中选择55个NSOC多发家系,对每个家庭中2个或3个NSOC患者进行WES,结果发现唇腭裂患者共有的ADAMTS9基因罕见SNP位点(rs149253049)在印度3个家庭得到显著的结果(P=2×10-6)[12]。Liu等[15]对一个多发家系样本中的5名非综合征型CP患者进行WES研究,发现ARHGAP29基因是非综合征型CP的一个重要候选基因,在颌面部组织中高度表达,并用多个功能试验证实了这一结论。通过小鼠整体原位杂交和免疫荧光染色研究发现,ARHGAP29基因在唇腭的形成中发挥重要作用[25]。Fu等[18]对来自56个家系的115名研究对象进行WES研究发现53个区域存在一个或多个与NSOC发生风险有关的罕见杂合缺失。Liu等[13]对8名NSCL/P胎儿进行WES研究,发现并验证了16个基因位点与NSCL/P发生风险有关。
3. NSOC目标区域测序研究:WGS理论上讲是遗传变异定位的金标准,而全外显子组测序则针对全基因组内编码蛋白质的外显子进行检测,但即便应用NGS,上述方法仍需耗费大量时间,成本高昂且数据分析困难。正因如此,成本较低的目标区域测序成为目前广泛采用的、研究罕见变异的方法。
Leslie等[14]选择GWAS和其他研究中发现的13个阳性区域,在1 409个亚洲和欧洲核心家系中进行目标区域测序研究,研究发现PAX7、FGFR2和NOG基因多个功能性位点与非综合征型CL/P的发生风险有关。PAX7基因突变在体外测定中破坏了编码的转录因子的DNA折叠,继而影响唇腭裂的发生[14]。动物实验表明,PAX7参与神经嵴诱导,并在颅神经嵴细胞中表达,缺乏PAX7的小鼠会出现鼻和上颌骨结构畸形[26]。FGFR2基因在颌面部发育中发挥重要作用。NOG也是唇腭裂的重要候选基因,编码BMP拮抗剂的NOG的转录物主要在腭发育期间在上皮中表达[27]。Savastano等[17]在2016年对来自188个家系的206名研究对象进行目标区域测序,发现ARHGAP29基因上10个罕见突变位点可能和非综合征型CL/P的发生风险有关。Peng等[16]采用病例对照研究设计,选择GWAS研究中发现的18个NSOC阳性位点,对103名NSOC患者和100名健康对照进行目标区域测序,研究发现10个基因位点(ABCA4、MYH9、MTHFR、CRISPLD2、FGF8、PVRL1、FOXE1、VAX1、FGFR2和IRF6)上的22种非同义突变与NSCO发生风险有关。
三、二代测序数据的统计分析NGS产生的海量数据需要适当的方法进行分析。NGS数据分析主要通过特定的生物信息学软件来实现,分析过程通常基于以下步骤:①原始数据的初步处理,包括碱基质量校准、序列比对等,这一过程主要是完成原始数据的校准化处理,为后续分析提供标准的文件,Illumina Pipeline等生物信息工具主要用于NSOC测序数据的质量控制;②利用突变查找相关软件或工具比较原始的突变信息,主要包括SNP、Indel、CNV等,Burrows-Wheeler Aligner(BWA),the Genome Analysis Toolkit 1.6(GATK)和NNOVAR(Annotate Variation)等是常用于NSOC二代测序数据突变筛选的工具,而NCBI dbSNP 135和1 000 Genomes Project是常用的比对数据库;③针对原始的突变信息,利用额外的信息对原始突变信息进行过滤、筛选和优化,得到可用于分析的比较可靠的变异信息[28]。由于数量巨大且变异位点的低频率,应用传统关联分析的方法检验测序数据单个罕见位点的效应把握度过低。理论上讲,某个致病基因或通路内可存在多个罕见遗传位点影响疾病的发生风险,因此合并计算多个位点的效应将提高统计学效率。将单个罕见位点的效应进行加和及校正;采用折叠法将同一个功能元件上的变异合并;以及采用聚合法对罕见变异进行加权等方法都可用于后续突变效应的分析[29]。序列核关联性检验(sequence kernel association test,SKAT)常被用于NSOC等复杂疾病罕见遗传变异的分析[30]。
四、总结虽然NGS的发展实现了高通量、低成本、快速测序的目的,但NGS的测序准确性最高只能达到99.99%,根据不同的平台,NGS数据的错误率大概在百分之零点几至百分之几,微小的错误率也会造成大量的测序错误,因此,NGS技术仍需进一步完善以便更好地用于NSOC的病因探索。从GWAS到WES,再到WGS,产生的数据的数量急剧增加,因此在使用NGS数据进行疾病病因探索的过程中,庞大的数据量的清洁和处理是一项巨大的工程。NSOC的遗传危险因素研究主要采用家系研究设计,由于家系研究的特殊性,未来针对NSOC二代测序研究的数据处理工作仍需进一步深入探索。此外,如何合理地评估和比较NGS产生的突变位点是NGS分析中的另一重要问题,处理NGS数据将涉及个体几千万个罕见遗传变异,并且大多数观察到的遗传变异都位于未知的基因编码区域内,这在一定程度上限制了我们对这些遗传位点的研究。虽然各种研究设计都可以用于NSOC致病机制的探索,但对测序所发现的新发突变的生物学功能的解释仍是研究的难点。此外,面对NGS的海量信息,NSOC遗传危险因素研究需要开发可以用于处理任何数据集内多个致病位点的研究方法和策略,并有效地应用这些策略进行NSOC病因学的探索。
NGS提供了NSOC遗传危险因素探索的新思路和新方法,可以发现NSOC的罕见遗传变异、拷贝数变异等,更好地解释NSOC的遗传度。如何更好地规范NGS的测序流程,更有效地分析和储存NSOC的NGS数据以及其可能涉及的伦理学问题都是未来需要关注的热点。
利益冲突: 无
| [1] | Mossey PA, Little J, Munger RG, et al. Cleft lip and palate[J]. Lancet, 2009, 374(9703): 1773–1785. DOI:10.1016/S0140-6736(09)60695-4 |
| [2] | Leslie EJ, Marazita ML. Genetics of cleft lip and cleft palate[J]. Am J Med Genet C Semin Med Genet, 2013, 163C(4): 246–258. DOI:10.1002/ajmg.c.31381 |
| [3] | Mangold E, Ludwig KU, Nöthen MM. Breakthroughs in the genetics of orofacialclefting[J]. Trends Mol Med, 2011, 17(12): 725–733. DOI:10.1016/j.molmed.2011.07.007 |
| [4] | Stanier P, Moore GE. Genetics of cleft lip and palate:syndromic genes contribute to the incidence of non-syndromic clefts[J]. Hum Mol Genet, 2004, 13(Suppl 1): R73–81. DOI:10.1093/hmg/ddh052 |
| [5] | Dixon MJ, Marazita ML, Beaty TH, et al. Cleft lip and palate:understanding genetic and environmental influences[J]. Nat Rev Genet, 2011, 12(3): 167–178. DOI:10.1038/nrg2933 |
| [6] | Cooper ME, Ratay JS, Marazita ML. Asian oral-facial cleft birth prevalence[J]. Cleft Palate Craniofac J, 2006, 43(5): 580–589. DOI:10.1597/05-167 |
| [7] | Ludwig KU, Mangold E, Herms S, et al. Genome-wide Meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci[J]. Nat Genet, 2012, 44(9): 968–971. DOI:10.1038/ng.2360 |
| [8] | Leslie EJ, Carlson JC, Shaffer JR, et al. Genome-wide Meta-analyses of nonsyndromicorofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate[J]. Hum Genet, 2017, 136(3): 275–286. DOI:10.1007/s00439-016-1754-7 |
| [9] | Reich DE, Lander ES. On the allelic spectrum of human disease[J]. Trends Genet, 2001, 17(9): 502–510. DOI:10.1016/S0168-9525(01)02410-6 |
| [10] | Collins FS, Guyer MS, Chakravarti A. Variations on a theme:cataloging human DNA sequence variation[J]. Science, 1997, 278(5343): 1580–1581. DOI:10.1126/science.278.5343.1580 |
| [11] | Bureau A, Parker MM, Ruczinski I, et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts[J]. Genetics, 2014, 197(3): 1039–1044. DOI:10.1534/genetics.114.165225 |
| [12] | Bureau A, Younkin SG, Parker MM, et al. Inferring rare disease risk variants based on exact probabilities of sharing by multiple affected relatives[J]. Bioinformatics, 2014, 30(15): 2189–2196. DOI:10.1093/bioinformatics/btu198 |
| [13] | Liu YP, Xu LF, Wang Q, et al. Identification of susceptibility genes in non-syndromic cleft lip with or without cleft palate using whole-exome sequencing[J]. Med Oral Patol Oral Cir Bucal, 2015, 20(6): e763–770. DOI:10.4317/medoral.20758 |
| [14] | Leslie EJ, Taub MA, Liu H, et al. Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci[J]. Am J Hum Genet, 2015, 96(3): 397–411. DOI:10.1016/j.ajhg.2015.01.004 |
| [15] | Liu H, Busch T, Eliason S, et al. Exome sequencing provides additional evidence for the involvement of ARHGAP29 in Mendelianorofacialclefting and extends the phenotypic spectrum to isolated cleft palate[J]. Birth Defects Res, 2017, 109(1): 27–37. DOI:10.1002/bdra.23596.2016 |
| [16] | Peng HH, Chang NC, Chen KT, et al. Nonsynonymous variants in MYH9 and ABCA4 are the most frequent risk loci associated with nonsyndromicorofacial cleft in Taiwanese population[J]. BMC Med Genet, 2016, 17: 59. DOI:10.1186/s12881-016-0322-2 |
| [17] | Savastano CP, Brito LA, Faria áC, et al. Impact of rare variants in ARHGAP29 to the etiology of oral clefts:role of loss-of-function vs missense variants[J]. Clin Genet, 2017, 91(5): 683–689. DOI:10.1111/cge.12823 |
| [18] | Fu J, Beaty TH, Scott AF, et al. Whole exome association of rare deletions in multiplex oral cleft families[J]. Genet Epidemiol, 2017, 41(1): 61–69. DOI:10.1002/gepi.22010 |
| [19] | Stranneheim H, Wedell A. Exome and genome sequencing:a revolution for the discovery and diagnosis of monogenic disorders[J]. J Intern Med, 2016, 279(1): 3–15. DOI:10.1111/joim.12399 |
| [20] | Royer-Bertrand B, Rivolta C. Whole genome sequencing as a means to assess pathogenic mutations in medical genetics and cancer[J]. Cell Mol Life Sci, 2015, 72(8): 1463–1471. DOI:10.1007/s00018-014-1807-9 |
| [21] | Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants[J]. Proc Natl Acad Sci USA, 2015, 112(17): 5473–5478. DOI:10.1073/pnas.1418631112 |
| [22] | Beaty TH, Marazita ML, Leslie EJ. Genetic factors influencing risk to orofacial clefts:today's challenges and tomorrow's opportunities[J]. F1000 Res, 2016, 5: 2800. DOI:10.12688/f1000research.9503.1 |
| [23] | Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder[J]. Nat Genet, 2010, 42(1): 30–35. DOI:10.1038/ng.499 |
| [24] | Kerrigan JJ, Mansell JP, Sengupta A, et al. Brown N, Sandy JR. Palatogenesis and potential mechanisms for clefting[J]. J R Coll Surg Edinb, 2000, 45(6): 351–358. |
| [25] | Leslie EJ, Mansilla MA, Biggs LC, et al. Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22[J]. Birth Defects Res A Clin Mol Teratol, 2012, 94(11): 934–942. DOI:10.1002/bdra.23076 |
| [26] | Mansouri A, Stoykova A, Torres M, et al. Dysgenesis of cephalic neural crest derivatives in Pax7-/-mutant mice[J]. Development, 1996, 122(3): 831–838. |
| [27] | He FL, Xiong W, Wang Y, et al. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis[J]. Dev Biol, 2010, 347(1): 109–121. DOI:10.1016/j.ydbio.2010.08.014 |
| [28] |
高彧辉. 基于二代测序数据的SNP发现策略及其初步应用[D]. 南昌: 南昌大学, 2013. DOI: 10.7666/d.Y2402520.
Gao YH. SNP calling and its preliminary application based on next generation sequencing data[D]. Nanchang: Nanchang University, 2013. DOI: 10.7666/d.Y2402520. |
| [29] |
周家蓬, 裴智勇, 陈禹保, 等. 基于高通量测序的全基因组关联研究策略[J]. 遗传, 2014, 36(11): 1099–1111.
Zhou JP, Pei ZY, Chen YB, et al. Strategies of genome-wide association study based on high-throughput sequencing[J]. Hereditas, 2014, 36(11): 1099–1111. DOI:10.3724/SP.J.1005.2014.1099 |
| [30] | Wu MC, Lee S, Cai TX, et al. Rare-variant association testing for sequencing data with the sequence kernel association test[J]. Am J Hum Genet, 2011, 89(1): 82–93. DOI:10.1016/j.ajhg.2011.05.029 |