 2017, Vol. 38
2017, Vol. 38文章信息
- 张佳峰, 姚佳明, 范钦, 陈婉君, 潘晓红, 丁晓贝, 杨介者, 傅涛.
- Zhang Jiafeng, Yao Jiaming, Fan Qin, Chen Wanjun, Pan Xiaohong, Ding Xiaobei, Yang Jiezhe, Fu Tao.
- 浙江省义乌市2016年新报告HIV/AIDS的HIV-1亚型和传播簇分析
- Analysis on HIV-1 subtypes and transmission clusters in newly reported HIV/AIDS cases in Yiwu, Zhejiang Province, 2016
- 中华流行病学杂志, 2017, 38(12): 1688-1693
- Chinese journal of Epidemiology, 2017, 38(12): 1688-1693
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2017.12.021
-
文章历史
收稿日期: 2017-06-18




2. 325035 温州医科大学检验医学院 生命科学学院;
3. 322000 义乌市疾病预防控制中心
2. School of Laboratory Medicine and Life Science, Wenzhou Medical University, Wenzhou 325035, China;
3. Yiwu Municipal Center for Disease Control and Prevention, Yiwu 322000, China
分子流行病学在研究HIV流行分布和追踪流行变迁等方面发挥了关键作用[1]。近年来国际上提出对不同地域和不同高危人群进行HIV传播动力学和传播网络分析,及时发现快速成簇的毒株,针对这些病毒特征的HIV感染者给予特别关注,实施“簇内监测、干预、治疗”有效干预方式[1-3]。本研究选择在流动人口众多、艾滋病疫情上升风险较大的义乌市(中国商贸代表地区)开展分子流行病学研究,以期掌握该地区HIV流行毒株亚型分布特点,并进一步分析传播簇特征,为制定针对性干预措施提供切实建议和科学依据。
对象与方法1.研究对象:为浙江省义乌市168例2016年新报告的HIV/AIDS病例,占当年新报告且现住址为义乌市病例的93.9%(168/179),其余11例因未能采血或留存血样未纳入研究。EDTA抗凝全血(6~8 ml)由当地CDC在对病例进行首次CD4+T淋巴细胞(CD4)检测时采集,并及时分离血浆,定期送至浙江省CDC,样本置于-80 ℃保存备用。所有病例采样前均未接受过抗病毒治疗。本研究通过了浙江省CDC伦理委员会的伦理学审查,实施过程严格遵守相关的伦理学标准。
2.研究方法:
(1)RNA提取和PCR扩增:使用苏州天隆生物科技有限公司的病毒RNA/DNA提取试剂盒结合其NP968核酸提取仪提取RNA,以此为模板,采用文献[4]所述方法先后进行RT-PCR和巢式PCR扩增HIV部分pol基因(包括蛋白酶全长和反转录酶前300个氨基酸位点),扩增片段长度为1 316 bp。
(2)PCR产物测序:扩增产物用1%的琼脂糖凝胶电泳分析,将含目的片段的产物送往杭州擎科梓熙生物技术有限公司进行纯化和测序。
(3)亚型分析:使用Sequencher 5.0软件对测序结果进行拼接整理,使用BioEdit 7.2.0软件将整理好的样本序列同国际参考株序列进行Clustal W比对和校正,国际参考株序列来自美国Los Alamos国家实验室的HIV序列数据库(https://www.hiv.lanl.gov)。用Mega 6.0软件构建Neighbor-Joining系统进化树(选择Kimura 2-parameter模型,Bootstrap= 1 000),并计算毒株间的遗传距离。通过样本序列同国际参考株聚集情况初步判断样本亚型,进一步使用在线分析工具COMET(https://comet.lih.lu/index.php)复核[5]。对于未能与已知的亚型和流行重组型(circulating recombinant form,CRF)聚集的序列考虑为独特重组型(unique recombinant form,URF),采用RIP(www.hiv.lanl.gov/content/sequence/RIP/RIP.html)和jpHMM重组分析工具(http://jphmm.gobics.de)分析重组类型及断点。
(4)同源性分析:利用HIV序列数据库的HIV BLAST工具对毒株序列进行同源性分析[6],寻找同源性最高的前10条序列,根据其注解的地理位置,推测所分析毒株可能的来源地。
(5)传播簇分析:本研究定义的传播簇为构建的系统进化树中簇内病例数≥2个,成簇的Bootstrap≥90%,且簇内平均基因距离≤0.015。
(6)耐药分析:使用美国斯坦福大学HIV耐药数据库中的CPR 6.0在线软件工具(http://cpr.stanford.edu/cpr.cgi)分析基因型耐药情况[7],根据WHO 2009版监测性耐药突变(surveillance drug resistance mutation,SDRM)位点确定是否存在SDRM突变[8]。
3.统计学分析:使用SPSS 19.0软件进行χ2检验,以P<0.05为差异有统计学意义。
结果1.人口学特征:168例研究对象以男性为主(86.3%),年龄(39.1±13.4)岁,汉族占绝大多数(96.4%),外省户籍居多(66.7%),婚姻状况以未婚(43.5%)和已婚(42.9%)为主,教育程度以初中及以下居多(67.9%),职业分布较广,以农民、工人和民工最多(44.6%),其次是商业服务(29.2%);感染途径为同性传播(51.2%)和异性传播(48.8%)。见表 1。

2.亚型分析:168例样本共有151例(89.9%)成功获得序列。经分析共发现9种亚型,分别为CRF01_AE(74例,49.0%),CRF07_BC(64例,42.4%),CRF08_BC(5例,3.3%),CRF55_01B(3例,2.0%),B、CRF45_cpx、CRF59_01B和CRF85_BC各1例,还发现1例URF(B/C),见图 1。HIV亚型的人群分布见表 1。利用HIV BLAST对新CRF(指近几年新出现或者传入的CRF)和独特重组型进行同源性分析(表 2),结果显示可能的来源地较多,与其户籍地匹配的仅2例(JH20167466、JH20167316)。

|
| 注:●国际参考株;▼同性传播;◇异性传播;粗线为传播簇;a携带PI耐药突变M46L;b携带NRTI耐药突变F77L;c携带NNRTI耐药突变Y181C;节点处仅显示≥70%的Bootstrap值 图 1 2016年义乌市新报告HIV/AIDS的HIV系统进化树分析 |
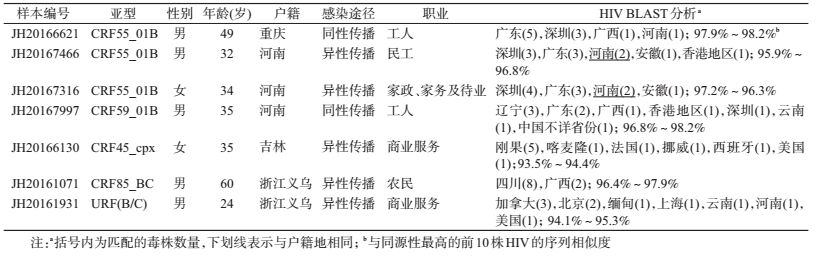
3. URF重组分析:样本JH20161931与现有报道的所有亚型和流行重组型均未能聚集,考虑为独特重组型,经RIP和jpHMM重组分析工具分析为B和C亚型的重组,序列长度1 195 bp,相对于HXB2的位置为2 253→3 447,是由B亚型(2 253→3 229)和C亚型(3 230→ 3 447)重组构成,重组断点的间隔区位置为3 212±31(图 2)。流行病学调查显示该病例为24岁男性,义乌本地户籍,有商业、非商业、非婚异性性接触史,自述与8人有非婚性行为。HIV BLAST同源性分析显示该毒株与加拿大、缅甸和美国,以及北京、上海、云南和河南地区报道的毒株存在较高同源性,序列相似性为94.1%~95.3%(表 2)。

|
| 注:A. jpHMM分析得出的重组结果;B.分析片段(带背景色)的基因图谱,其中数值表示相对于HXB2的位置 图 2 l例HIV独特重组型的重组分析 |
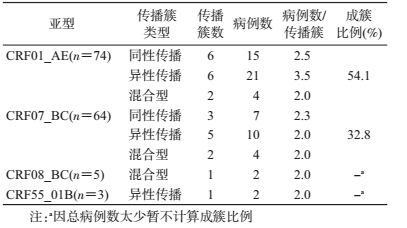
4.传播簇分析:本研究共发现26个传播簇(含65个病例),总成簇比例为43.0%(65/151)。其中CRF01_AE成簇比例为54.1%(40/74),高于CRF07_BC的32.8%(21/64)(χ2=6.278,P=0.016);异性传播感染者的成簇比例为50.6%(38/75),同性传播感染者成簇比例为35.5%(27/76),两者差异无统计学意义(χ2=3.529,P=0.060)。各传播簇大小(传播簇包含的病例数)介于2~5例/簇之间,平均为2.5例/簇,其中CRF01_AE的传播簇平均大小为3.3例/簇,高于CRF07_ BC的2.1例/簇。CRF01_AE中异性传播的传播簇平均大小最大,达到3.5例/簇。所有病例感染途径均为异性传播的传播簇占46.2%(12/26),均为同性传播的占34.6%(9/26),包括异性/同性传播(即混合型)的占19.2%(5/26)。见图 1和表 3。
5.传播性耐药结果:共发现7例存在SDRM,总耐药传播率为4.6%(7/151),处于耐药传播的低度流行水平(<5%);携带SDRM的病例均只携带1个耐药突变,共有3种不同类型,分别为PI耐药突变M46L(5/151,3.3%),NRTI耐药突变F77L(1/151,0.7%)和NNRTI耐药突变Y181C(1/151,0.7%)。此外还发现携带M46L耐药突变的4例处于CRF01_AE的同一个传播簇内(图 1),该4例均为男性,异性传播感染。
讨论HIV亚型多样性对于艾滋病流行具有多方面的影响,包括病毒致病性、传播、诊断、治疗和疫苗研制等[9]。义乌市是目前全球最大的小商品集散地,流动人口规模庞大,艾滋病流行危险因素广泛存在。本研究显示当地流行毒株以CRF01_AE和CRF07_BC为主(占91.4%),多种亚型并存,也发现新CRF和URF;多种传播簇并存且特征多样,CRF01_AE毒株和异性传播感染者的成簇比例较高,这些结果能为当地制定靶向干预措施提供参考依据。
本研究发现义乌市的优势亚型与2013年杭州市新报告病例的优势亚型相似[4],也与上海市以CRF01_AE和CRF07_BC为主的组成类似[10]。除优势亚型外,本研究还发现一些近年来国内新出现的CRF。CRF55_01B起源于深圳市MSM,且在当地有一定的流行(9.2%,99/1 072)[11],在全国已呈散发态势[12]。CRF55_01B在浙江省MSM人群已有报道[13],本研究又检出3例,印证其全国散发流行状况。本研究发现的3例中1例为同性传播,2例为异性传播(双阳夫妻),提示CRF55_01B可通过双性MSM的“桥梁”作用传播到异性人群中。本研究还发现1例CRF59_01B(其最早于2013年在辽宁省MSM人群中被鉴定发现[14]),1例CRF85_BC(于2016年在四川省异性传播人群被鉴定发现[15]),以及1例URF(B/C),说明新的CRF随着人口流动已在省际间传播,也提示HIV重组仍在不断产生,将继续产生新的CRF和URF,从而使HIV亚型种类更加多样和复杂,其对艾滋病疫情会产生深远影响。义乌市出现的新CRF和URF(表 2)通过HIV BLAST分析显示可能的来源地较多,说明这些毒株传播较为复杂,需要进行细致的流行病学调查。由于商贸地区人口组成复杂,流动人口来源广,不同亚型间交叉容易产生新重组,新重组亚型很容易通过流动人口进行跨省传播,需要进一步强化亚型监测。本研究发现的1例CRF45_cpx在浙江省首次发现,国内也未见报道,通过HIV BLAST分析推测来自非洲。新的复杂重组的传入会对当地艾滋病流行产生深远影响,鉴于义乌市外籍商贸人数较多(常住外商1.3万人),应开展对外籍人群的重点筛查和预防干预工作。
传播簇体现的是直接或间接传播的流行病学关联,如若干HIV感染者毒株基因序列相近,则他们可能通过某些传播事件相关联[16]。义乌市CRF01_AE毒株成簇比例较高,说明该毒株在当地有相对较多的遗传紧密集群,意味其有更高的HIV传播和扩散风险。义乌市异性传播感染者的成簇比例高达50.6%(38/75),说明HIV在当地通过异性传播途径的风险较高;相比而言,同性传播感染者成簇比例(35.5%)稍低,这可能与当地MSM因社会歧视和担心名誉受损选择远离居住地,而在更广泛的地域范围内活动有关[13, 17]。本研究发现80.8%的传播簇所含病例感染途径均相同,说明同性传播和异性传播存在各自相对独立的流行圈,但也发现19.2%为混合型(包括同性/异性传播),说明HIV毒株通过MSM的双性行为与异性人群交叉渗透,也印证中国MSM人群存在相当比例的双性性行为[18]。这些传播簇研究结果有助于分析当地疫情传播特征,为确定重点干预对象提供参考信息。分析这些传播簇的流行特征并加强重点干预,可预防传播簇的进一步扩张,有望控制和减少艾滋病新发感染。
本研究发现义乌市新病例中耐药传播整体处于低水平(<5%),携带SDRM的7例病例均只携带单一耐药突变,暂不会对现有抗病毒治疗措施产生不利影响。耐药突变具有相对稳定性,有些可携带超过10年[19],因此可作为传播流行的“示踪器”。本研究发现4例携带有PI耐药突变M46L处于CRF01_AE的同一个传播簇内,进一步印证相互间的传播关系。
本研究只在局部地区开展横断面研究,无法动态分析传播簇的变化,也未能全面掌握感染毒株来源,应进一步结合个案的流行病学调查全面分析其传播网络特征,为该地区制定针对性艾滋病防治措施提供参考依据。
利益冲突: 无
| [1] |
汪宁, 钟平.
中国HIV分子流行病学30年[J]. 中华流行病学杂志, 2015, 36(6): 541–546.
Wang N, Zhong P. Molecular epidemiology of HIV in China:1985-2015[J]. Chin J Epidemiol, 2015, 36(6): 541–546. DOI:10.3760/cma.j.issn.0254-6450.2015.06.001 |
| [2] | Little SJ, Pond SLK, Anderson CM, et al. Using HIV networks to inform real time prevention interventions[J]. PLoS One, 2014, 9(6): e98443. DOI:10.1371/journal.pone.0098443 |
| [3] | Wang XC, Wu YS, Mao L, et al. Targeting HIV prevention based on molecular epidemiology among deeply sampled subnetworks of men who have sex with men[J]. Clin Infect Dis, 2015, 61(9): 1462–1468. DOI:10.1093/cid/civ526 |
| [4] | Zhang JF, Guo ZH, Yang JZ, et al. Genetic diversity of HIV-1 and transmitted drug resistance among newly diagnosed individuals with HIV infection in Hangzhou, China[J]. J Med Virol, 2015, 87(10): 1668–1676. DOI:10.1002/jmv.24223 |
| [5] | Struck D, Lawyer G, Ternes AM, et al. COMET:adaptive context-based modeling for ultrafast HIV-1 subtype identification[J]. Nucleic Acids Res, 2014, 42(18): e144. DOI:10.1093/nar/gku739 |
| [6] | Mendoza Y, Bello G, Castillo Mewa J, et al. Molecular epidemiology of HIV-1 in Panama:origin of non-B subtypes in samples collected from 2007 to 2013[J]. PLoS One, 2014, 9(1): e85153. DOI:10.1371/journal.pone.0085153 |
| [7] | Gifford RJ, Liu TF, Rhee SY, et al. The calibrated population resistance tool:standardized genotypic estimation of transmitted HIV-1 drug resistance[J]. Bioinformatics, 2009, 25(9): 1197–1198. DOI:10.1093/bioinformatics/btp134 |
| [8] | Bennett DE, Camacho RJ, Otelea D, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance:2009 update[J]. PLoS One, 2009, 4(3): e4724. DOI:10.1371/journal.pone.0004724 |
| [9] | Aldrich C, Hemelaar J. Global HIV-1 diversity surveillance[J]. Trends Mol Med, 2012, 18(12): 691–694. DOI:10.1016/j.molmed.2012.06.004 |
| [10] |
吴健, 王绪琴, 周艳秋, 等.
上海市2007-2013年HIV-1分子流行病学演变趋势[J]. 中国艾滋病性病, 2016, 22(1): 5–9.
Wu J, Wang XQ, Zhou YQ, et al. Molecular epidemiological characteristics of human immunodeficiency virus type 1 in Shanghai, 2007-2013[J]. Chin J AIDS STD, 2016, 22(1): 5–9. DOI:10.13419/j.cnki.aids.2016.01.03 |
| [11] | Zhao J, Cai W, Zheng C, et al. Origin and outbreak of HIV-1 CRF55_01B among MSM in Shenzhen, China[J]. J Acquir Immune Defic Syndr, 2014, 66(3): e65–67. DOI:10.1097/QAI.zhlxbxzz-38-12-168800144 |
| [12] | Han X, Takebe Y, Zhang W, et al. A Large-scale Survey of CRF55_01B from Men-Who-Have-Sex-with-Men in China:implying the Evolutionary History and Public Health Impact[J]. Sci Rep, 2015, 5: 518147. DOI:10.1038/srep18147 |
| [13] |
张佳峰, 杨介者, 潘晓红, 等.
浙江省男男性行为人群HIV-1新近感染者的毒株亚型和传播簇研究[J]. 中华流行病学杂志, 2015, 36(1): 61–66.
Zhang JF, Yang JZ, Pan XH, et al. HIV-1 subtype diversity and transmission clusters among men having sex with men who recently got HIV-l infection, in Zhejiang province[J]. Chin J Epidemiol, 2015, 36(1): 61–66. DOI:10.3760/cma.j.issn.0254-6450.2015.01.015 |
| [14] | Han X, An M, Zhang W, et al. Genome sequences of a novel HIV-1 circulating recombinant form (CRF59_01B) identified among men who have sex with men in Northeastern China[J]. Genome Announc, 2013, 1(3): e00315-13. DOI:10.1128/genomeA.00315-13 |
| [15] | Su L, Wei DB, Yang H, et al. Identification of a Novel HIV-1 circulating recombinant form (CRF85_BC) in Sichuan, China[J]. AIDS Res Hum Retroviruses, 2016, 32(9): 895–899. DOI:10.1089/AID.2016.0053 |
| [16] | Grabowski MK, Redd AD. Molecular tools for studying HIV transmission in sexual networks[J]. Curr Opin HIV AIDS, 2014, 9(2): 126–133. DOI:10.1097/COH.zhlxbxzz-38-12-168800040 |
| [17] | Chen M, Ma YL, Su YZ, et al. HIV-1 genetic characteristics and transmitted drug resistance among men who have sex with men in Kunming, China[J]. PLoS One, 2014, 9(1): e87033. DOI:10.1371/journal.pone.0087033 |
| [18] | Yun K, Xu JJ, Reilly KH, et al. Prevalence of bisexual behaviour among bridge population of men who have sex with men in China:a meta-analysis of observational studies[J]. Sex Transm Infect, 2011, 87(7): 563–570. DOI:10.1136/sextrans-2011-050079 |
| [19] | Martínez-Prats L, Luczkowiak J, Gutiérrez F, et al. Sustained profile of transmitted drug resistance mutations for more than 10 years in a HIV type 1-infected patient[J]. AIDS Res Hum Retroviruses, 2011, 27(1): 41–45. DOI:10.1089/aid.2010.0104 |