 2017, Vol. 38
2017, Vol. 38文章信息
- 陈海霞, 蔡超, 刘静仪, 张治国, 原梅, 贾俊楠, 孙照刚, 黄海荣, 高基民, 李卫民.
- Chen Haixia, Cai Chao, Liu Jingyi, Zhang Zhiguo, Yuan Mei, Jia Junnan, Sun Zhaogang, Huang Hairong, Gao Jimin, Li Weimin.
- 不同可变数目串联重复序列组合对中国流行结核分枝杆菌分辨力的评价研究
- Discriminatory power of variable number on tandem repeats loci for genotyping Mycobacterium tuberculosis strains in China
- 中华流行病学杂志, 2017, 38(6): 794-799
- Chinese Journal of Epidemiology, 2017, 38(6): 794-799
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2017.06.021
-
文章历史
收稿日期: 2016-10-27




2. 101149 北京市结核病胸部肿瘤研究所北京市结核病耐药重点实验室;
3. 101149 北京, 首都医科大学附属北京胸科医院国家结核病临床实验室;
4. 102200 北京市昌平区结核病防治所检验科;
5. 044000 山西省运城市急救中心检验科;
6. 100069 北京, 首都医科大学公共卫生学院临床流行病学北京市重点实验室
2. The Drug-resistant TB Key Laboratory of Beijing, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing 101149, China;
3. National Tuberculosis Clinical Laboratory of China, Beijing Chest Hospital, Capital Medical University, Beijing 101149, China;
4. The Institute of Tuberculosis Prevention and Control of Changping District, Beijing 102200, China;
5. Yuncheng City Emergency Center, Yuncheng 044000, China;
6. Beijing Municipal Key Laboratory of Clinical Epidemiology, School of Public Health, Capital Medical University, Beijing 100069, China
据统计,2014年我国结核病(tuberculosis,TB)分别有930 000例新发病例和38 000例死亡病例,TB例数居世界第三,是我国所面临的一项严峻公共卫生问题[1]。结核基因分型是分子流行病学的核心技术。目前国际上使用较为广泛的结核分枝杆菌(M. tuberculosis,MTB)基因分型方法主要包括:可变数目串联重复序列(variable number of tandem repeats,VNTR)以及目前逐渐被广泛使用的长序列多态性(large sequence polymorphism,LSP)、单核苷酸多态性(single nucleotide polymorphism,SNP)和全基因组测序。其中,全基因组测序作为一项新兴技术,因其快速、准确、高效的优点,在结核病基础研究中发挥了重要作用[2-4];但是其昂贵的价格以及复杂的分析方法使之在我国的结核病预防控制领域广泛推广尚需时日。VNTR分型技术因其操作简便,分析快速以及便于跨实验室比较等优点,对暴发流行、近期传播、疫情监测等结核流行病学实际工作具有指导意义。目前国际上使用较广泛的有15-VNTR及24-VNTR[5-6]。但VNTR分型技术具有同源异质性的缺陷。因此采用LSP和SNP先行谱系定义,再行VNTR分型是目前MTB基因分型的一种高效策略。Luo等[7]曾在采用SNP鉴定北京谱系,及其“现代”和“古老”亚谱系MTB的基础上,评价了几个对其分辨率较高的VNTR位点,并以此为工具在我国开展了卓有成效的分子流行病学研究工作[8-9]。但是除北京谱系外,我国还有约40%的其他谱系MTB的流行,有必要找出一套适合我国结核病预防控制的最优VNTR分型方案。
本研究对2007年全国结核病耐药性基线调查收集的31省4 116株MTB 15-VNTR基因分型,汉高指数(Hunter-Gaston Index,HGI)评估各地区VNTR位点的分辨力,并根据分辨力及遗传稳定性排列分析,为各省推出一套高效的VNTR位点组合,筛选出适合我国国情的MTB基因分型策略,并将其应用于结核病预防控制的实际工作。
材料与方法1.材料:
(1)实验菌株:2007年全国结核病耐药性基线调查涵盖中国31个省(自治区、直辖市),共收集4 116株MTB。经鉴定均为MTB[10-11]。标准菌株H37Rv由北京胸科医院国家结核病临床实验室提供。
(2)主要试剂和仪器:2×Taq Master Mix、100 bp Ladder均购自北京康为世纪生物科技有限公司;15-VNTR位点引物序列参照文献[6],由北京擎科生物公司合成;Bio Rad凝胶成像仪以及Quantity One软件购自美国Bio Rad生命科学公司。
2.实验室方法:
(1)DNA提取:① 煮沸法:吸取200 μl冻存菌液于无菌EP管,100 ℃水浴20~30 min,12 000 r/min离心4 min,上清液即含DNA。② 酚-氯仿法:对于扩增失败的菌株,吸取50 μl冻存菌液接种于中性罗氏培养基斜面,37 ℃恒温培养4周;待长出菌落,用接种环轻轻刮取细菌,100 ℃,30 min灭活。标准酚-氯仿法提取。
(2)VNTR基因分型:采用标准的15-VNTR方法。PCR扩增条件:94 ℃预变性5 min;94 ℃变性30 s,65 ℃退火30 s,72 ℃延伸45 s,30~35个循环;延伸72 ℃ 10 min;PCR扩增产物由1.2%琼脂糖凝胶分离,Bio-Rad凝胶成像仪系统上处理分析,100 bp DNA Ladder作为参照。
3.分析方法:
(1)样本收集质控:复苏或PCR扩增失败(经过重复2次PCR扩增有≥5个VNTR位点失败)的菌株不纳入本研究。
(2)实验室质控:实验设立对照菌株H37Rv在同一VNTR位点的扩增片段大小是否相同。从检测标本中随机挑10个菌株,每株不同VNTR位点重复检测3次,比较每次扩增片段是否一致。
(3)结果分析:Quantity one软件分析每个VNTR位点的PCR结果,H37Rv为参照标准。重复单元数目公式:各VNTR位点重复单位数目=(PCR产物片段长度-VNTR位点侧翼片段长度)/重复单元片段长度。
(4)HGI计算:计算各VNTR位点以及不同VNTR位点组合的分辨力[12]。公式:
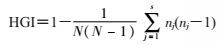
式中N代表样本菌株总数;S代表基因型总数;nj代表拥有第j种基因型的菌株总数。
由于不同地区同一VNTR位点偶尔存在极端值的情况,因此将VNTR位点HGI的中位数值(M)用来估算其对全国范围内MTB的总分辨率。
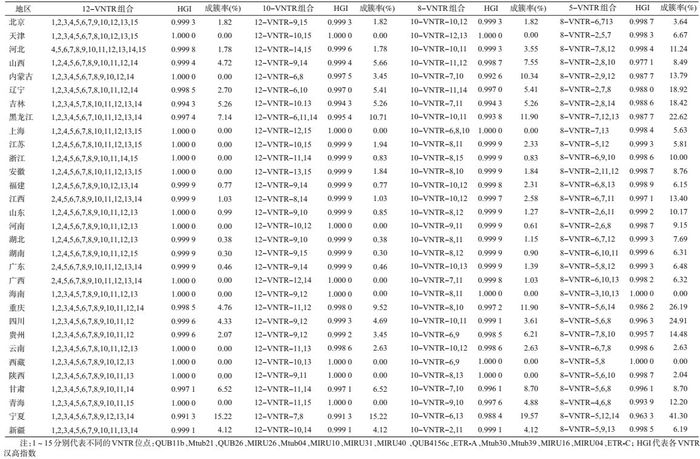
(5)VNTR优化组合:针对每个省份MTB谱系流行特征,依据参考文献[13]提示谱系不同,VNTR分辨力不同。15个VNTR位点根据分辨率不同划分为高、中、低3个水平,高、中分辨率且稳定性好的VNTR位点作为一线候选位点,分辨率低(HGI>0.1)但稳定性好的位点作为二线候选位点,分辨率高但稳定性差(该地区扩增失败的菌株数超过该地区总菌株数的5%)位点则舍弃。并以15-VNTR的分辨率及成簇率为参考,为每个省份均分别排列推出12-VNTR、10-VNTR、8-VNTR、5-VNTR[14]。并采用HGI和成簇率进行评估,成簇率=(成簇的菌株数-成簇基因型数)/总菌株数。
结果1.纳入分析菌株及重复性:
(1)菌株收集:2007年全国结核病耐药性基线调查所收集的4 116株MTB,150株因年久缺失、复苏或PCR扩增失败,最终3 996株MTB纳入本研究。
(2)检测结果的重复性:对照菌株H37Rv在同一VNTR位点的扩增片段大小相同。每株MTB在不同VNTR位点的3次重复检测结果均完全一致。
2. 15位点VNTR分辨率和稳定性:所有菌株的分型结果提示:QUB26、QUB11b、Mtub21、MIRU16位点扩增失败的菌株数量较多,比例分别为11.85%(470/3 996)、2.67%(106/3 996)、3.08%(122/3 996)、4.06%(122/3 996),提示这些位点相对其他VNTR位点遗传稳定性较差。并发现不同地区同一VNTR位点稳定性不同,同一地区不同VNTR位点遗传稳定性也不同(表 1)。其中QUB26在广西、广东、湖南、上海、宁夏、山东等13省份扩增失败的菌株数超过5%。QUB11b在广西、广东、江西、河北4省份扩增失败的菌株数超过5%。Mtub21在山西、河北、江西3省扩增失败的菌株数超过5%。MIRU16在湖南、浙江、山西、青海4省扩增失败的菌株数超过5%。比较各个VNTR位点的HGI中值发现:QUB11b、Mtub21、QUB26、MIRU26、Mtub04、MIRU10、MIRU31、MIRU16位点HGI中值分别高达0.834、0.762、0.760、0.817、0.757、0.704、0.731、0.662(表 2),属于高分辨率位点(HGI>0.6);MIRU40、ETR-A、Mtub30、QUB4156c、MIRU04、Mtub39属于中分辨率位点(0.3≤HGI≤0.6),ETR-C属于低分辨率位点(HGI<0.3)[15]。


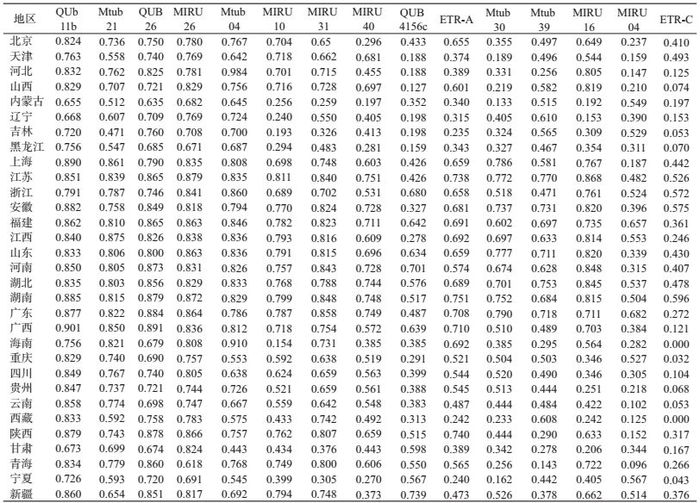
3.各省最佳VNTR组合位点分析:31个省份的15-VNTR分辨率均>0.994 3,成簇率均低于10%,其中除去重庆、贵州、江西、宁夏、甘肃,新疆、吉林、辽宁、黑龙江9个省份外,22个省份分辨率和成簇率均分别1和0。VNTR位点在不同省份的分辨率存在不同程度的差异,MIRU10、MIRU40、ETR-A、Mtub30、QUB4156c、MIRU04、MtUB39位点在个别省份分辨率过低,而ETR-C分辨率则是在全国普遍偏低。高分辨率位点QUB11b在某些省份稳定性较差。因此本研究为每省分别筛选12-VNTR、10-VNTR、8-VNTR、5-VNTR的4套组合(表 4)。发现QUB26位点被纳入西部地区VNTR组合明显高于东、中部地区,二线候选位点ETR-C被纳入南方地区VNTR组合高于北方地区,相邻地区间各VNTR组合通常是相同或仅有1个VNTR位点的差别。内蒙古、重庆、黑龙江地区选用10-VNTR作为最优组合,其他省份可用8-VNTR作为最优组合。
基因分型技术能够用于研究结核的暴发、近期传播、克隆扩张以及区分内源性复燃和外源性再感染,因此是结核病防控的重要手段。发达国家早在20世纪90年代就建有大型的MTB基因型数据库,在原有的暴发流行和接触调查的基础上开展成簇调查,以发现基因型相匹配的患者(成簇)之间的流行病学联系。所以结核病近期传播必须要有两个先决条件:① 菌株基因形成簇;② 患者有流行病学联系。因此本研究构建涵盖31个省3 996株MTB的15-VNTR数据库,有助于MTB传染源全国范围的追踪和簇调查。
目前我国虽已有多个不同VNTR组合评估的研究,但均仅针对某一谱系或某一地区,尚无多个谱系MTB流行的国家层面综合评估。本研究分析15个VNTR位点的分辨力发现,QUB11b、Mtub21、QUB26、MIRU26、Mtub04、MIRU10和MIRU31位点分辨率较高,与Comas等[13]和Chen等[16]研究结果一致。同时发现QUB26、MIRU16、Mtub21、QUB11b位点在部分省份遗传稳定性差。我国是多个MTB谱系流行且分布不均衡的结核病高负担国家。为提高结核病预防控制的效率,有必要向各省推荐一套优化的VNTR位点组合。优先推荐QUB11b、Mtub21、QUB26、MIRU26、Mtub04、MIRU10、MIRU31这7个较高分辨率位点,并以此为基础的12-VNTR、10-VNTR、8-VNTR 3套VNTR组合。
目前全基因组测序技术因居高的价格尚未应用于结核病的预防控制,纵然其价格下降,其以系统进化和群体遗传学的分析也是深奥的。鉴于VNTR基因分型同源异质性指数(homoplasy index,HI)较Spoligotyping更接近SNP[13],在谱系定义(LSP和SNP)基础上结合VNTR分型便是这一时期最高效的基因分型工具。因此对一个地区MTB进行全方位的VNTR基因分型并建立数据库,可利用成簇率和簇即代表近期的传播的概念,从时间和空间监测结核病疫情的变化。一个地区成簇率高,则疫情发展需密切关注,如成簇率低,则疫情平稳。根据Wirth等[17]以及之前研究结果提示[18-19],利用LSP、SNP以及VNTR数据研究MTB的群体遗传结构特征及系统进化,例如研究MTB的最近共祖年代、系统进化等,从时间和空间探讨该地区MTB的流行特征。总之,各省的结核病工作者可利用本研究推荐的VNTR组合建立当地结核菌基因分型数据库(含LSP或SNP鉴定和VNTR分型)进行疫情监测、初步系统进化和群体遗传学的分析,为当地结核病疫情控制策略的制定提供科学依据。
志谢: 感谢国家结核病参比实验室赵雁林教授等开展的2007-2008年度全国结核病耐药性基线调查;感谢国家结核病临床实验室霍凤敏实习研究员、姜广路副主任技师等为该工作提供的菌株资源和数据资源。利益冲突: 无
| [1] | WHO. Global tuberculosis report 2016[R/OL]. Geneva:WHO, 2016. http://www.who.int/tb/publications/global_report/en/. |
| [2] | Luo T, Comas I, Luo D, et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese[J]. Proc Natl Acad Sci USA, 2015, 112(26): 8136–8141. DOI:10.1073/pnas.1424063112 |
| [3] | Zhang HT, Li DF, Zhao LL, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance[J]. Nat Genet, 2013, 45(10): 1255–1260. DOI:10.1038/ng.2735 |
| [4] | Gardy JL, Johnston JC, Sui SJH, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak[J]. N Engl J Med, 2011, 364(8): 730–739. DOI:10.1056/NEJMoa1003176 |
| [5] | Supply P, Lesjean S, Savine E, et al. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units[J]. J Clin Microbiol, 2001, 39(10): 3563–3571. DOI:10.1128/JCM.39.10.3563-3571.2001 |
| [6] | Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis[J]. J Clin Microbiol, 2006, 44(12): 4498–4510. DOI:10.1128/JCM.01392-06 |
| [7] | Luo T, Yang CG, Gagneux S, et al. Combination of single nucleotide polymorphism and variable-number tandem repeats for genotyping a homogenous population of Mycobacterium tuberculosis Beijing strains in China[J]. J Clin Microbiol, 2012, 50(3): 633–639. DOI:10.1128/JCM.05539-11 |
| [8] | Luo T, Yang CG, Pang Y, et al. Development of a hierarchical variable-number tandem repeat typing scheme for Mycobacterium tuberculosis in China[J]. PLoS One, 2014, 9(2): e89726. DOI:10.1371/journal.pone.0089726 |
| [9] | Luo T, Yang CG, Gao Q. Mycobacterial interspersed repetitive-unit locus PCR amplification and Beijing strains of Mycobacterium tuberculosis[J]. J Clin Microbiol, 2011, 49(11): 4026–4027. DOI:10.1128/JCM.05389-11 |
| [10] | Zhao YL, Xu SF, Wang LX, et al. National survey of drug-resistant tuberculosis in China[J]. N Engl J Med, 2012, 366(23): 2161–2170. DOI:10.1056/NEJMoa1108789 |
| [11] | Pang Y, Zhou Y, Zhao B, et al. Spoligotyping and drug resistance analysis of Mycobacterium tuberculosis strains from national survey in China[J]. PLoS One, 2012, 7(3): e32976. DOI:10.1371/journal.pone.0032976 |
| [12] | Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems:an application of Simpson's index of diversity[J]. J Clin Microbiol, 1988, 26(11): 2465–2466. |
| [13] | Comas I, Homolka S, Niemann S, et al. Genotyping of genetically monomorphic bacteria:DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies[J]. PLoS One, 2009, 4(11): e7815. DOI:10.1371/journal.pone.0007815 |
| [14] | Kremer K, Arnold C, Cataldi A, et al. Discriminatory power and reproducibility of novel DNA typing methods for Mycobacterium tuberculosis complex strains[J]. J Clin Microbiol, 2005, 43(11): 5628–5638. DOI:10.1128/JCM.43.11.5628-5638.2005 |
| [15] | Sola C, Filliol I, Legrand E, et al. Genotyping of the Mycobacterium tuberculosis complex using MIRUs:association with VNTR and spoligotyping for molecular epidemiology and evolutionary genetics[J]. Infect Genet Evol, 2003, 3(2): 125–133. DOI:10.1016/S1567-1348(03)00011-X |
| [16] | Chen YY, Chang JR, Huang WF, et al. Genetic diversity of the Mycobacterium tuberculosis Beijing family based on SNP and VNTR typing profiles in Asian countries[J]. PLoS One, 2012, 7(7): e39792. DOI:10.1371/journal.pone.0039792 |
| [17] | Wirth T, Hildebrand F, Allix-Béguec C, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex[J]. PLoS Pathog, 2008, 4(9): e1000160. DOI:10.1371/journal.ppat.1000160 |
| [18] |
丁朋举, 惠明, 梁倩, 等.
中国北方部分地区北京谱系结核分枝杆菌群体遗传关系的分析[J]. 中华流行病学杂志, 2013, 34(4): 374–378.
Ding PJ, Hui M, Liang Q, et al. Origin, phylogeny, and spread of Mycobacterium tuberculosis Beijing lineage in the five provinces of northern China[J]. Chin J Epidemiol, 2013, 34(4): 374–378. DOI:10.3760/cma.j.issn.0254-6450.2013.04.015 |
| [19] |
李颖, 付育红, 原梅, 等.
四川盆地结核分枝杆菌群体遗传学特征研究[J]. 中华流行病学杂志, 2015, 36(4): 374–378.
Li Y, Fu YH, Yuan M, et al. Study on the population-genetics of Mycobacterium tuberculosis from Sichuan Basin in China[J]. Chin J Epidemiol, 2015, 36(4): 374–378. DOI:10.3760/cma.j.issn.0254-6450.2015.04.017 |