 2016, Vol. 37
2016, Vol. 37文章信息
- 刘红, 高晓艳, 付士红, 李铭华, 翟友刚, 孟维珊, 孙肖红, 王环宇, 吕志, 申辛欣, 曹玉玺, 何英, 梁国栋 .
- Liu Hong, Gao Xiaoyan, Fu Shihong, Li Minghua, Zhai Yougang, Meng Weishan, Sun Xiaohong, Wang Huanyu, Lyu Zhi, Shen Xinxin, Cao Yuxi, He Ying, Liang Guodong .
- 基于第12节段基因序列的版纳病毒分子遗传进化分析
- Analysis on molecular genetic evolution of Banna virus based on the 12th segment
- 中华流行病学杂志, 2016, 37(9): 1277-1282
- CHINESE JOURNAL OF EPIDEMIOLOGY, 2016, 37(9): 1277-1282
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2016.09.019
-
文章历史
收稿日期: 2016-03-07




2. 102206 北京, 中国疾病预防控制中心病毒病预防控制所脑炎室
2. Department of Virual Encephalitis, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 102206, China
版纳病毒(Banna virus,BAV)是呼肠孤病毒科东南亚十二节段双链RNA病毒属(Seadornavirus genus)的模式病毒,其基因组为12节段双链RNA病毒[1]。自1987年首次从我国云南西双版纳脑炎患者标本中分离以来,相继在位于北温带、亚热带以及热带(赤道至北纬42°)的中国[2]、印度尼西亚(印尼)[3]、越南[4]等地的猪[5]、牛、蜱[6-7]、蠓及3属10种蚊虫标本中分离[8]。2013年,在匈牙利巴拉顿湖(Lake Balaton)的淡水鲤鱼肠内容物[9]和中国云南省蚊虫标本中[10],分别分离到版纳病毒样病毒(Banna-like virus,BALV),表明BAV蕴含着极丰富基因多样性已分化出新的变种并且具有跨地域传播的能力。鉴于BAV为新发虫媒病毒且与人畜疾病有密切关系。了解该病毒在自然界的起源进化过程,无论对于该病毒病的预防控制以及对于理解新发现的双链RNA病毒的分子遗传特性等病毒学理论研究均有重要意义。本研究对1980-2012年从不同地域和宿主中分离的BAV第12节段基因序列开展了系统的起源和进化分析,该节段编码双链RNA结合蛋白,是基因组中最保守也是目前该病毒基因序列信息最丰富的一个节段,因此采用此节段开展分析可以很好的了解该病毒在自然界进化的全貌。
材料与方法1. 病毒分离及其序列测定:自2000年以来,在我国各地的猪场、牛场、鱼塘以及农村居民房屋等生境采集蚊虫标本。标本经过分类鉴定和登记后分装在冻存管中,置于液氮罐中保存,干冰运回中国CDC病毒性脑炎室开展病毒分离鉴定工作。病毒毒株的分离、复苏、扩增、RNA的提取、cDNA的制备过程同文献[8]。使用覆盖BAV第12节段整个编码区的引物(BAV12F:AAA TTG ATA GYG YTT GCG TAA GAG,BAV12R:GTT CTA AAT TGG ATA CYG CGT GC)进行PCR 扩增。PCR产物送生工生物工程(上海)有限公司进行序列测定。BAV第12节段基因序列采用ATGC软件进行拼接、编辑、校正。
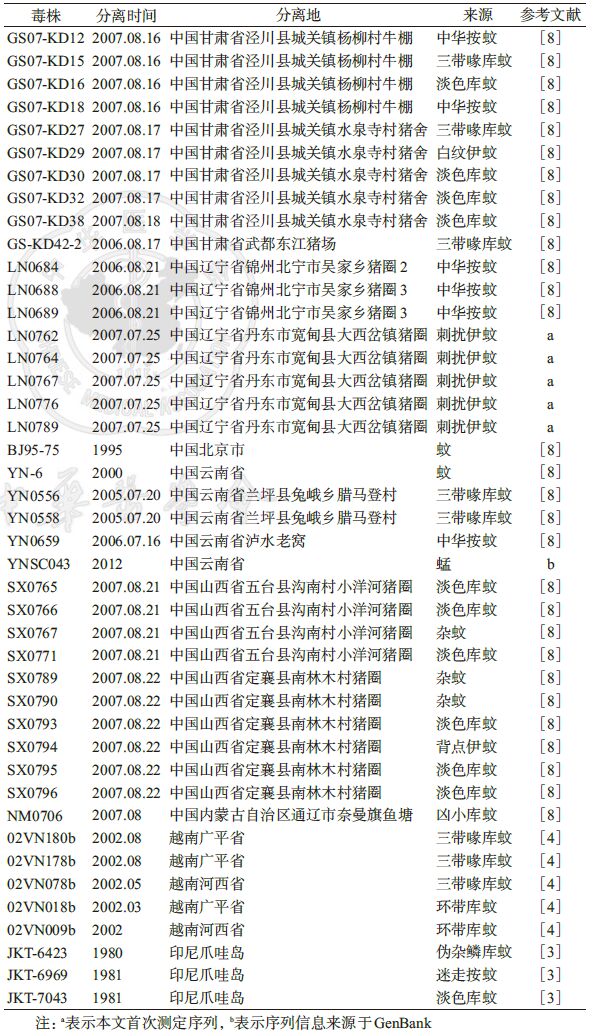
2. BAV第12节段基因序列数据集:对1980-2012年世界各地分离到的44株BAV第12节段基因序列信息进行了分析,毒株包括我国7个省或地区蚊虫和蜢标本中分离的35株病毒;1987年首次在云南省脑炎病例分离的病毒株(BAV_ch),以及3株印尼蚊虫标本分离株(JKT6423、JKT6969、JKT7043)和5株越南蚊虫标本分离株(02VN009b、02VN018b、02VN078b、02VN178b、02VN180b)。这些毒株分离自3属10种蚊虫标本,1种蜢标本以及病例标本(表 1)。 BAV分布在赤道至北纬42°之间,包括赤道地区(印尼)属于热带雨林气候;亚热带地区(越南和中国云南省)属于热带季风性气候;以及北温带地区(中国内蒙古、辽宁、山西、北京、甘肃)属典型的中温带季风性气候。2013年及2015年分别在匈牙利(北纬47°)以及中国云南省报道分离到BALV,为了阐明这些毒株和BAV真正的亲缘关系,本研究同时构建了BAV与BALV株序列数据集,包括2株从GenBank下载的BALV序列和44株BAV第12节段序列信息。
3. BAV第12节段序列分析:BAV数据集的序列比对和编辑分别采用ClustalW 2和BioEdit软件进行;核苷酸和氨基酸同源性基因以及其序列特征分别使用MegAlign(DNASTAR,Madison,WI,USA)、GenDoc软件分析,系统进化分析采用Mega 6.0软件进行。
4. 基于时间尺度的BAV系统进化分析:进行基于时间尺度的系统进化分析前,采用SIMPLOT和 RDP4 软件包开展病毒数据集的重组信息的筛查,未在病毒第12节段检测到重组序列信号。BAV第12节段的最近共同进化祖先(time to the most recent common ancestor,tMRCA)、碱基替换速率以及具有时间尺度的系统进化分析采用BEAST 软件包(http://beast.bio.ed.ac.uk/)进行[11]。采用MrModelTest软件进行最佳碱基替换模型的筛选,GTR+I+G模型被评估为最适合的碱基替换模型。分别选择严格分子钟(strict clock)和松弛分子钟(relaxed clock)匹配4种种群增长模型进行计算参数设置。运算链长根据预实验结果以最终ESS数值>200为标准进行调整。
结 果1. 基于时间尺度的BAV系统进化分析:贝叶斯种群动态模型被评估为最适合的参数模型,据此模型计算获得BAV最大可信树主要进化分支的后验概率数值均>0.8,表明结果具有很高的信度。进一步分析发现,BAV最近共同进化祖先出现在距今315年前(95% HPD:63~619),之后逐渐演化为两个主要的基因型别,基因A型和基因B型。基因B型主要由赤道地区分离株构成,其最近共同进化祖先出现时间在距今45(95% HPD:36~81)年前;基因A型主要由北纬15°~42°区域分离的毒株构成,出现时间为距今217(95% HPD:51~424)年前。基因A型之后又经历了两次种群分化事件,分别在距今92年前和164年前根据南北地域差异分化出了基因A1(毒株主要分布于北纬30°以上)和基因A2亚型(毒株主要分布于北纬15°~30°之间)。BAV的系统聚类表现出明显的地域分布特征,即同一地域分离的BAV毒株聚类成簇,表现出更近的亲缘关系,地理距离远的毒株进化距离较远聚类成不同的进化分支(图 1)。

|
| 图 1 具有时间尺度的BAV系统进化树 |
由北纬30°以上分离株组成的A1 亚型混杂着3株越南株(图 1,黑色正方形标注),而由北纬15°~30°分离株组成的基因A2亚型混杂着5株2007年从中国辽宁地区分离的BAV(图 1,黑色三角形标注)。
不同吸血昆虫及不同宿主动物来源的BAV系统聚类分析显示,BAV依据地域分布聚类成簇,不同宿主来源的病毒在进化树的分布没有表现出明显的依据种属来源不同而聚类成簇的现象。
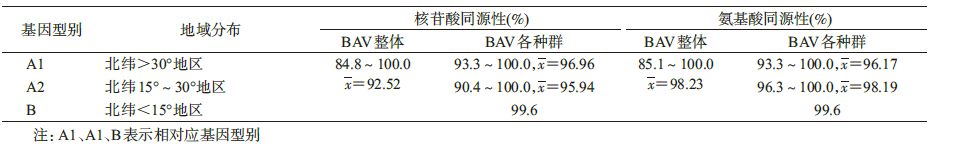
2. BAV基因序列分析:对1980-2012年不同宿主和不同地域分离的44株BAV第12基因节段的核苷酸序列分析发现:①基因结构特征:BAV第12节段的编码区长度为624 nt,编码208个氨基酸。不同基因型别、不同宿主来源的病毒编码区长度不存在差异。44株BAV的5′末端序列很保守,均为5′ACC AAC ATG GAT CCT; 3′末端的序列为TCC GCG TAA(TGA) AYA R。②BAV的终止密码子的使用具有明显的基因型别特征:基因A1亚型,基因A2亚型及基因B型的终止密码子分别为TGA、TAA和TGA。③核苷酸和氨基酸同源性分析:44株BAV核苷酸同源性在84.8%~100.0%之间,核苷酸同源性平均为92.52%;氨基酸同源性在85.1%~100.0%之间,平均氨基酸同源性为98.23%。位于北纬30°以上的 A1亚型毒株之间核苷酸同源性在93.3%~100.0%之间,μ=96.96%;氨基酸同源性在93.3%~100.0%之间,平均为96.17%;位于北纬15°~30° A2亚型毒株之间的核苷酸同源性在90.4%~100.0%之间,平均为95.94%,氨基酸同源性在96.3%~100.0%之间,平均为98.19%;北纬15°以下印尼分离株群基因B型的核苷酸和氨基酸的同源性为99.6%(表 2)。同一地域分离的毒株之间同源性可达到99.0%,而最南部的毒株与最北端的毒株核苷酸同源性仅有84.8%;云南蚊虫、蜢和患者标本中分离的毒株之间同源性为 99.7%,表现出密切的亲缘关系。
3. BAV与BAV样病毒的亲缘关系分析:为了详细阐明新分离BALV与BAV及其他Seadornavirus属病毒的亲缘关系。采用最大似然法构建了系统进化树(图 2),结果显示匈牙利和中国云南省新分离的两株BALV隶属于Seadornavirus属,位于BAV、辽宁病毒(Liaoning virus,LNV)和卡迪皮诺病毒(Kadipiro virus,KDV)进化分支之间,两株BALV聚类成簇,形成了与BAV有密切亲缘关系的独立进化分支。

|
| 图 2 BAV与其他东南亚十二节段双链RNA病毒属病毒亲缘关系 |
4. 进化速率、选择压力分析:BAV进化速率计算结果显示其平均碱基替换速率为2.33×10-3(95% HPD:2.84×10-4~8.52×10-3)碱基替换/位点/年。BAV第12节段选择压力分析结果显示,其非同义/同义置换率(ω=dN/dS ratio)ω=0.157±0.053。
讨 论呼肠孤病毒科Seadornavirus属是国际上新成立病毒属,目前包括3种病毒分别为BAV、LNV和KDV[12]。BAV是该病毒属代表种也是国际上第一个由我国科学家分离并以我国地名命名的病毒[13]。该病毒中文名称曾经为新环状病毒、Colti病毒等[12],BAV最初分离自不明原因发热和病毒性脑炎病例标本,同时检测到病例抗体滴度呈现4倍以上增高,因此被认为是一个新发现的与人类脑炎有密切关联的虫媒病毒。本研究从分子进化角度对BAV起源进化、碱基替换速率、毒株之间的亲缘关系、宿主屏障等科学问题进行了分析。结果显示BAV最早的共同进化祖先大约出现在300多年前,与其他已知的虫媒病毒相比属于新发病毒:如乙型脑炎病毒的共同进化祖先出现时间约为距今3 000多年[14],LNV的共同进化祖先为1 500年前出现[15]。值得注意的是最近在巴西等地流行并造成严重公共卫生问题的寨卡病毒(Zika Virus,ZIKV)共同祖先出现的时间为距今325 年前[15],提示BAV与ZIKA是在同一时期出现的虫媒病毒,这类新发的虫媒病毒出现年代不长,进化变异迅速、生存适应能力强,可被多种宿主媒介携带,通过叮咬人畜造成疾病的传播,是一类需要给予足够重视的新发病原体,应加强对这类新发虫媒病毒的基础和应用研究以应对可能突发的重大公共卫生事件。
BAV的毒株之间具有明显的地域分布特征,亲缘关系与毒株之间的地理距离成正比,但不同基因型别的毒株可以通过季风和候鸟的作用传播至新的地域[7-8],迅速适应并进化出在适合当地生存的种群,表明BAV不存在地理屏障。不同宿主种类分离的BAV无论从基因结构、核苷酸和氨基酸同源性还是系统进化特征分析均不具有种属差异。这些特性进一步说明,BAV可以适应不同的宿主和生存环境,具有极强的传播能力和生存适应性。
BAV第12节段的选择压力ω<1,表明其在自然界主要受净化选择压力作用,即大部分有害的突变被净化选择压力清除。BAV在产生的初期由“旧病毒”演化而来,因此积累了丰富的种群多样性以高效的适应环境和宿主。值得注意的是,近期不断有BAV变种在不同地域和宿主中分离的报道,如匈牙利新分离的BALV[9]和中国云南省新分离的芒市病毒[10]等均提示BAV的活动异常活跃,其变异已突破病毒种内的限制而进化出新的病毒种,这些新病毒种是否仍然维持原有BAV生物学表型和致病力,或者出现完全不同生物学特性,以至于突破原有宿主攻击范围造成新的公共卫生危机,是亟待研究的课题。
本研究表明BAV不具备地域和宿主屏障,快速的突变率使其准种中蕴藏着丰富的基因多样性,同时赋予其丰富的分化潜力,为了适应新的宿主和生境该病毒可能会产生新的基因型别甚至是与“旧版纳病毒”差异明显的“新型版纳病毒变种”,特别是BAV及其衍生病毒的地域分布已超出我们以往的认知,具有进一步扩大的倾向。鉴于BAV与人畜疾病密切的关系特别是其具有活跃的变异和传播能力,应加强世界范围特别是北半球高纬度地区传播媒介和人畜动物中的监测。
但需要指出的是,BAV为12节段双链RNA病毒,虽然其第12节段基因序列最为保守且其序列信息最丰富可以代表BAV进化特征。但要真正全面了解BAV在自然界进化状态,亟需进一步开展基于其全基因组序列进化分析。
| [1] | Fauquet CM, Mayo MA, Maniloff J.Virus taxonomy:Ⅷth report of the international committee on taxonomy of viruses[M].San Diego: Elsevier Academic Press, 2005: 504–510 |
| [2] | 徐普庭, 王逸民, 左建民, 等. 从云南省无名热病人和脑炎病人分离到新环状病毒[J]. 病毒学报 , 1990, 6 (1) : 27–33 P T, Wang YM, Zuo JM, et al. New orbiviruses isolated from patients with unknown fever and encephalitis in Yunnan province[J]. Chin J Virol , 1990, 6 (1) : 27–33 |
| [3] | Brown SE, Gorman BM, Tesh RB, et al. Coltiviruses isolated from mosquitoes collected in Indonesia[J]. Virology , 1993, 196 (1) : 363–367 DOI:10.1006/viro.1993.1490 |
| [4] | Nabeshima T, Nga PT, Guillermo P, et al. Isolation and molecular characterization of Banna virus from mosquitoes,Vietnam[J]. Emerg Infect Dis , 2008, 14 (8) : 1276–1279 DOI:10.3201/eid1408.080100 |
| [5] | 徐普庭, 王逸民, 左建民, 等. 云南省猪牛体内发现人源同型新环状病毒[J]. 病毒学报 , 1990, 6 (4) : 327–331 P T, Wang YM, Zuo JM, et al. Recovery of the same type of virus as human new orbivirus from sera of Cattles and pigs collected in Yunnan Province[J]. Chin J Virol , 1990, 6 (4) : 327–331 |
| [6] | 李其平, 谢杏初, 郅琦, 等. 首次从新疆的蜱和病牛血清分离到新环状病毒[J]. 地方病通报 , 1992, 7 (4) : 64–69 Q P, Xie XC, Zhi Q, et al. First isolation of new orbivirus (Banna) from ticks and infected cattle sera in Xinjiang[J]. Endemic Dis Bull , 1992, 7 (4) : 64–69 |
| [7] | Liu H, Gao XY, Liang GX. Newly recognized mosquito-associated viruses in mainland China,in the last two decades[J]. Virol J , 2011, 8 : 68 DOI:10.1186/1743-422X-8-68 |
| [8] | Liu H, Li MH, Zhai YG, et al. Banna virus,China,1987-2007[J]. Emerg Infect Dis , 2010, 16 (3) : 514–517 DOI:10.3201/eid1603.091160 |
| [9] | Reuter G, Boros á, Delwart E, et al. Novel seadornavirus (family Reoviridae) related to Banna virus in Europe[J]. Arch Virol , 2013, 158 (10) : 2163–2167 DOI:10.1007/s00705-013-1712-9 |
| [10] | Wang JL, Li HC, He YW, et al. Isolation and genetic characterization of Mangshi virus:a newly discovered seadornavirus of the reoviridae family found in Yunnan Province,China[J]. PLoS One , 2015, 10 (12) : e0143601 DOI:10.1371/journal.pone.0143601 |
| [11] | Drummond AJ, Rambaut A. BEAST:Bayesian evolutionary analysis by sampling trees[J]. BMC Evol Biol , 2007, 7 : 214 DOI:10.1186/1471-2148-7-214 |
| [12] | Attoui H, Jaafar FM, de Micco P, et al. Coltiviruses and seadornaviruses in North America,Europe,and Asia[J]. Emerg Infect Dis , 2005, 11 (11) : 1673–1679 DOI:10.3201/eid1111.050868 |
| [13] | Tao SJ, Chen BQ. Studies of coltivirus in China[J]. Chin Med J , 2005, 118 (7) : 581–586 |
| [14] | Gao XY, Liu H, Li MH, et al. Insights into the evolutionary history of Japanese encephalitis virus (JEV) based on whole-genome sequences comprising the five genotypes[J]. Virol J , 2015, 12 : 43 DOI:10.1186/s12985-015-0270-z |
| [15] | Faye O, Freire CCDM, Iamarino A, et al. Molecular evolution of Zika virus during its emergence in the 20th century[J]. PLoS Negl Trop Dis , 2014, 8 (1) : e2636 DOI:10.1371/journal.pntd.0002636 |