 2015, Vol. 36
2015, Vol. 36文章信息
- 王铖, 戴俊程, 孙义民, 谢兰, 潘良斌, 胡志斌, 沈洪兵.
- Wang Cheng, Dai Juncheng, Sun Yimin, Xie Lan, Pan Liangbin, Hu Zhibin, Shen Hongbing.
- 遗传风险评分的原理与方法
- Genetic risk score:principle,methods and application
- 中华流行病学杂志, 2015, 36(10): 1062-1064
- Chinese Journal of Epidemiology, 2015, 36(10): 1062-1064
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2015.10.005
-
文章历史
- 投稿日期: 2015-06-15




2. 博奥生物集团有限公司;
3. 清华大学医学院医学系统生物学研究中心;
4. 博奥颐和健康科学技术有限公司
2 Capital Bio Corporation;
3 Medical Systems Biology Research Center, Tsinghua University School of Medicine;
4 Capital Bio eHealth Corporation
近十年来,随着人们对基因组认识的不断加深以及技术不断改革进步,遗传关联研究尤其是全基因组关联研究(GWAS)作为流行病学研究的重要方法,取得了巨大成功。目前为止,已有越来越多样本量大、可靠性好的遗传关联研究发表在世界顶尖期刊上,揭示了大量的疾病易感位点,为从遗传水平进行疾病风险预测奠定了基础。风险评分(risk score)是流行病学研究中评价风险预测能力的重要方法之一[1],纳入遗传易感因素进行风险评分,从而评价遗传易感因素在风险预测模型中的效果的方法称为遗传风险评分(genetic risk score,GRS)。
遗传风险评分方法主要有5种:简单相加遗传风险评分(a simple count genetic risk score,SC-GRS)[2];以OR值作为权重的遗传风险评分(an odds ratio weighted genetic risk score,OR-GRS)[2, 3, 4];直接基于logistic回归的遗传风险评分(a direct logistic regression genetic risk score,DL-GRS)[5];多基因遗传风险评分(a polygenic genetic risk score,PG-GRS)[5];可释方差遗传风险评分(explained variance weighted genetic risk score,EV-GRS)。本文介绍其计算方法以及进行应用举例,为了方便计算过程的描述,方法中假设涉及到的遗传易感位点相互之间不存在连锁不平衡,如不加注释,一般以相加模型作用于疾病,所有参数估计均使用logistic回归模型。方法涉及的公式中,以D表示疾病状态(D=1表示样本为病例;D=0表示样本为健康对照),以G表示一组遗传易感位点风险等位基因数的集合向量(Gi表示第i个遗传易感位点的风险等位基因的数量)。
1. SC-GRS:是最简单的GRS方法,其计算方法不涉及任何单核苷酸多态性(SNP)效应的先验信息,即为所有SNP的风险等位基因数量的和(公式1),相关的疾病模型见公式(2)。

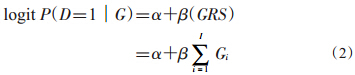
该方法通俗易懂,计算简单,因此在早期研究中应用较多,尤其是在SNP效应不能稳定估计的时候更为适用[6, 7]。但是,此方法假设所有SNP对疾病具有相同效应,该假设在现实研究中几乎不可能存在,因此,在建立疾病风险预测模型研究中很少使用。
2. OR-GRS:相比于SC-GRS,该方法考虑SNP对疾病的不同效应,以SNP效应作为权重,计算所有纳入模型SNP的OR值权重和(公式3、4),其相关的疾病模型如公式(5)所示。



为预先确定固定权重,实际应用中,往往使用大样本量、可靠性好的研究(如Meta分析)中对数转化后的单风险等位基因OR值作为权重。该方法中具有较大OR值的SNP对疾病风险贡献更大。其假设更为合理,因此被广泛应用于疾病风险模型预测的研究中[8],但因其估计依赖外部信息,不适用于一些不能准确估计SNP效应的研究。随着GWAS兴起,大量发现的疾病易感位点均运用该方法纳入遗传风险预测的研究中。
3. DL-GRS:该方法类似OR-GRS,但是基于的权重来自于已有原始数据,利用这些数据拟合logistic回归模型,以模型中估计的SNP效应作为权重,计算所有纳入模型SNP的OR值权重和公式(6),其相关的疾病模型如公式(7)所示。


该方法仅依赖现有数据,不需要外部研究的OR值作为权重,但是随之而来的问题即是该评分用于外部数据的可靠性有待商榷。该方法常常应用于无法通过外部信息准确估计SNP效应的研究。但是当该评分应用于另一个独立的数据时,其拟合的效果往往不如其在建立该评分的数据中拟合的效果。因此,研究者往往会设置两个或多个阶段的研究,以发现样本估计SNP效应,以独立验证样本进行验证[9, 10]。
4. PG-GRS:类似于DL-GRS,该方法依赖于现有数据。与以上GRS估计方法不同,该方法以哑变量的形式考虑每个SNP,即应用遗传模型中的共显性模型(公式8),其相关的疾病模型如公式(9)所示。
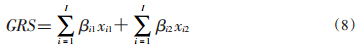
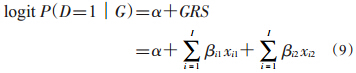
5. EV-GRS:是基于既往的风险评分方法,同时纳入考虑了SNP效应和最小等位基因频率(MAF)。除已经报道的SNP效应外,该方法在权重中增加了最小等位基因部分(公式10、11),其相关疾病模型如公式(11)所示。
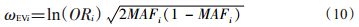

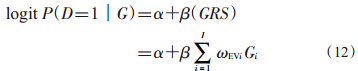
MAF可以来源于既往对应人群的公共数据库,如dbSNP、1 000 Genomes计划或者HAPMAP计划等。该方法认为,对于每个SNP,SNP效应和MAF均为衡量其对疾病贡献的重要因素,当OR值固定时,疾病风险将随着MAF增加而增加。该方法 在模拟数据中表现出了比较好的效果,但是尚无实际数据的应用评价结果,该遗传风险评分的效果有待进一步论证。
随着发现位点的增多,往往在一个研究中会纳入大量的位点进行评分,因此会增加模型的复杂性,从而产生过度拟合的情况,因此,一些研究在进行位点效应估计时,会采用惩罚回归模型[12](例如Lasso或者弹性网络等)或者机器学习的方法(例如支持向量机等)。这些方法尚未广泛使用,其应用效果有待后续研究的评估。
| [1] Shen HB,Jin GF. Genome-wide association study(GWAS) and risk prediction of complex disease:advances and prospects[J]. Chin J Epidemiol,2011,32(7):643-649. (in Chinese)沈洪兵,靳光付. 全基因组关联研究与复杂疾病风险预测的现状与展望[J]. 中华流行病学杂志,2011,32(7):643-649. |
| [2] Talmud PJ,Hingorani AD,Cooper JA,et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes:Whitehall Ⅱ prospective cohort study[J]. BMJ,2010,340:b4838. |
| [3] De Jager PL,Chibnik LB,Cui J,et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility:a weighted genetic risk score[J]. Lancet Neurol,2009,8(12):1111-1119. |
| [4] Karlson EW, Chibnik LB,Kraft P,et al. Cumulative association of 22 genetic variants with seropositive rheumatoid arthritis risk[J]. Ann Rheum Dis,2010,69(6):1077-1085. |
| [5] Carayol J,Tores F,König IR,et al. Evaluating diagnostic accuracy of genetic profiles in affected offspring families[J]. Stat Med,2010,29(22):2359-2368. |
| [6] Paynter NP,Chasman DI,Paré G,et al. Association between a literature-based genetic risk score and cardiovascular events in women[J]. JAMA,2010,303(7):631-637. |
| [7] Janipalli CS,Kumar MVK,Vinay DG,et al. Analysis of 32 common susceptibility genetic variants and their combined effect in predicting risk of type 2 diabetes and related traits in Indians[J]. Diabet Med,2012,29(1):121-127. |
| [8] Vaarhorst AAM, Lu YC,Heijmans BT, et al. Literature-based genetic risk scores for coronary heart disease:the Cardiovascular Registry Maastricht (CAREMA) prospective cohort study[J]. Circ Cardiovasc Genet,2012,5(2):202-209. |
| [9] Ripatti S,Tikkanen E,Orho-Melander M,et al. A multilocus genetic risk score for coronary heart disease:case-control and prospective cohort analyses[J]. Lancet,2010,376(9750):1393-1400. |
| [10] Wu JC,Pfeiffer RM,Gail MH. Strategies for developing prediction models from genome-wide association studies[J]. Genet Epidemiol,2013,37(8):768-777. |
| [11] Dudbridge F. Power and predictive accuracy of polygenic risk scores[J]. PLoS Genet,2013,9(3):e1003348. |
| [12] Kooperberg C,LeBlanc M,Obenchain V. Risk prediction using genome-wide association studies[J]. Genet Epidemiol,2010,34(7):643-652. |