 2015, Vol. 36
2015, Vol. 36文章信息
- 黄育北, 宋丰举, 陈可欣.
- Huang Yubei, Song Fengju, Chen Kexin.
- 乳腺癌全基因组关联研究的现状及其应用价值的分析方法
- Current status of genome-wide association studies (GWAS) on breast cancer and application values of single nucleotide polymorphisms identified from GWAS
- 中华流行病学杂志, 2015, 36(10): 1058-1061
- Chinese Journal of Epidemiology, 2015, 36(10): 1058-1061
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2015.10.004
-
文章历史
- 投稿日期: 2015-06-05




乳腺癌是全球女性发病率和死亡率最高的恶性肿瘤[1]。2012年全球女性新发乳腺癌167万例,死亡52万例。乳腺癌的疾病负担预期在发展中国家,包括中国在内,均有一个明显的增加趋势[1]。虽然随机对照研究系统综述结果显示:乳腺钼靶筛查后20年,相对于未筛查组,筛查组乳腺癌死亡率下降20%[2],但是由于钼靶筛查成本相对较高,即使是在经济水平相对较高的国家和地区,钼靶普查的卫生经济学评价也并未取得预期的成本效果[3, 4]。因此,寻找合适的高危人群进行乳腺癌筛查,即高危人群筛查策略成为乳腺癌防控领域关注的热点。全基因组关联研究(GWAS)为寻找合适的高危筛查策略提供了良好的基础。为此本文将阐述乳腺癌GWAS的现状,并基于GWAS发现的单核苷酸多态性(SNP)位点,探讨这些SNP的潜在应用价值。 一、乳腺癌GWAS现状
自2007年首篇乳腺癌GWAS报道以来,目前国内外共有34篇乳腺癌易感性相关GWAS报道,包括5篇亚洲人群的GWAS[5, 6, 7, 8, 9]。共报道了93个与乳腺癌易感性相关的SNP位点,其中9个SNP与中国女性乳腺癌发病风险相关。此外,有16个SNP虽然来自欧洲人群[10, 11, 12, 13, 14],但是经过大规模人群验证后,发现同样与中国女性乳腺癌发病风险相关(达到GWAS的显著性水准)[15]。因此,共有25个GWAS来源SNP与中国女性乳腺癌风险相关,排除2个SNP与其他SNP存在高度连锁不平衡(r2>0.8),共有23个独立的SNP位点与中国女性乳腺癌易感性相关,包括亚洲人群中初始发现的8个SNP位点:rs4951011、rs10474352、rs9485372、rs2046210、rs10822013、rs7107217、rs2290203、rs4784227;以及欧洲人群中初始发现的15个SNP位点:rs616488、rs4849887、rs16857609、rs4973768、rs6828523、rs10941679、rs1432679、rs9693444、rs6472903、rs1219648、rs10771399、rs17356907、rs1292011、rs2236007、rs17817449。 二、GWAS来源SNP的应用价值研究现状
GWAS来源SNP的潜在应用价值包括应用于人群危险分层,以及进一步应用于人群筛查和干预的价值[16]。目前已有一些研究[17, 18, 19, 20, 21, 22],在传统乳腺癌风险预测模型(基于初潮年龄、初次生育年龄、乳腺癌家族史和良性乳腺疾病史等危险因素)基础上,引入GWAS发现的SNP后,提高了对乳腺癌风险预测能力。如Wacholder等[17]研究结果提示:在由年龄及其他4个乳腺癌传统危险因素构建的乳腺癌风险预测模型基础上,加入10个SNP遗传信息之后,模型的曲线下面积(AUC)由58.0%上升至61.8%。但是这些研究绝大多数来自于单纯的理论研究或模型研究[17, 19, 20, 21],很少有在真实人群中探讨这些SNP的筛查价值,如对原有筛查方法的准确性(包括特异度等),以及对乳腺癌的检出率和筛查发现乳腺癌的肿瘤特征(包括早期癌的比例、淋巴结转移等)的影响。
此外,既往的研究多数采用的是截至当时相应人群中所有发现的SNP位点[17, 18, 20, 22],并未对其进行筛选。随着未来GWAS成本的降低,以及更多GWAS的开展,预期未来可发现更多与乳腺癌易感性相关的SNP位点。采用上述方法势必会造成大量社会资源浪费。相反,如果只是片面地采用少数与乳腺癌发病关联极强的候选基因(如BRCA1/BRCA2)SNP用于界定高危人群或进行人群筛查,势必会存在一定的遗漏或选择偏倚。因此,从卫生经济学角度考虑,如何客观选取数量合适的目标SNP用于人群筛查,成为后GWAS时代亟待解决的问题之一。 三、乳腺癌筛查目标SNP的选取及其应用价值的分析方法
如何从众多GWAS来源的SNP中选取可用于乳腺癌筛查的SNP相关研究较少,本文通过系统总结既往研究,归纳出如下方法,可以考虑用于筛选目标SNP,并分析其潜在应用价值。
1. 系统检索国内外GWAS以确定备选SNP。系统检索国内外所有发表的乳腺癌易感性相关GWAS,排除预后相关的GWAS,以及GWAS之后的验证研究。从所有报道的SNP位点中整理出特定人群乳腺癌易感性相关的SNP位点,排除高度连锁不平衡的SNP位点,保留下来的SNP位点即为备选SNP。
2. 计算备选SNP的遗传风险解释比例(PVi)。从备选SNP中筛选出目标SNP,首先应考虑具有更大公共卫生学意义的SNP。虽然人群归因风险(PAR)是评价公共卫生学价值的重要指标,但是对于发病率较低的疾病,以及人群变异频率也相对较低的SNP而言,Pharoah等[20]认为PAR很可能不够,而SNP对特定疾病综合遗传风险的PVi可更好地反映该SNP的公共卫生学价值。其中PVi的计算方法为

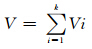
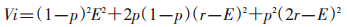

3. 基于PVi计算备选SNP对评价指标的影响。获得每个SNP的PVi之后,不能简单依靠PVi的排序大小来确定目标SNP。因为无法确定在引入新的SNP后,人群的风险分层是否会得到显著改善。因此,需要结合其他方法来评价在PVi最大SNP基础上,引入新的SNP是否能够改善人群的风险分层。
而传统的方法则是采用受试者工作特征曲线(ROC)的AUC以评价。但是目前研究发现,单一SNP通常很难引起AUC产生有统计学意义的改变,也即采用AUC来筛选目标SNP时,很可能遗漏很多有潜在价值的SNP[18]。因此,Pencina等[23]提出新的评价指标:整合区分指数(integrated discrimination improvement,IDI)和净再分类优化指数(net reclassification improvement,NRI)。
(1)IDI:其临床意义通常可解释为按照平均特异度,新方法对平均灵敏度的改善程度。而其统计学意义理论上等于2个预测模型的决定系数之差。按以下计算方法
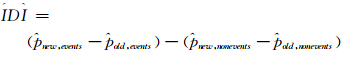
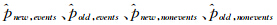 分别为新方法和原方法对患者的平均预测概率及新方法和原方法对非患者的平均预测概率。
分别为新方法和原方法对患者的平均预测概率及新方法和原方法对非患者的平均预测概率。
(2)NRI:其临床意义通常解释为新方法相对原方法,患者及非患者临床再分类的综合改善程度。风险分类上移视为优化;反之,视为减弱。计算方法为
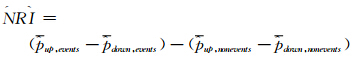
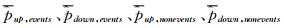 分别为新方法相对原方法,患者分类上移和下移的比例、非患者分类上移和下移的比例。
分别为新方法相对原方法,患者分类上移和下移的比例、非患者分类上移和下移的比例。
4. 比较备选SNP对AUC、IDI和NRI的影响,确定目标SNP。如上所述,AUC在发现目标SNP上,相对保守,与此相比,IDI和NRI更有可能发现对乳腺癌风险分层有改善的SNP[18]。如Mealiffe等[18]的研究结果提示,在传统Gail模型的基础上引入SNP 后,乳腺癌风险预测能力的总体AUC改善为3.7%。但是采用NRI评价在Gail模型基础上加入GWAS发现的SNP后,共计有8.5%的人群(5.6%的患者和2.9%的对照)风险分类会进一步得到改善。
在IDI和NRI两者结果的比较上,研究者通常需考虑①对于NRI,寻找有临床意义的风险分类标准(如Gail模型得出的5年绝对风险>1.5%)很难。针对该问题,目前已有统计学家提出了不受风险分类影响的NRI计算方法[24]。②由于IDI理论上等于2个预测模型的决定系数之差。因此,很容易受到样本量的影响,在样本量足够大时,即使IDI改变很小,也同样可能具有统计学意义。所以,如果同时通过IDI和NRI的改善以筛选目标SNP,相同样本量条件下,IDI可能筛选出更多的SNP。但以两者的临床意义而言,NRI相对更加明确。因此,研究者应该更多考虑通过NRI的改善来筛选适宜进行乳腺癌筛查的SNP。
简而言之,如果同时采用AUC、IDI和NRI预选目标SNP,而且备选SNP相对较多,同时资源亦相对充裕,可采用能同时显著改善AUC、IDI和NRI的SNP作为目标SNP。如果备选SNP数目相对有限,首先应该选取能够显著改善AUC的SNP,其次考虑能够显著改善NRI的SNP。在社会资源容许情况下,再考虑单独显著改善IDI的SNP。
5. 基于目标SNP,计算个体的遗传风险。确定目标SNP后,需要综合所有目标SNP的遗传变异情况,计算个体遗传风险分值(genetic risk score,GRS)的理论估计,如下式

6. 结合遗传及环境因素,确定合适的综合风险得分及分层方法。在得到个体GRS得分之后,仅提示个体的遗传易感基础,并不意味一定会发生特定疾病,因为环境因素是不可或缺的条件。因此必须同时整合GRS及环境危险因素,得到个体综合的风险得分,才可能真正发现适合的乳腺癌筛查高危人群。
2010年Zheng等[22]建立的中国女性乳腺癌传统因素风险预测模型中纳入的因素包括初潮年龄、初次生育年龄、腰臀比、乳腺癌家族史和良性乳腺疾病史。但该模型是基于病例对照研究构建的,其中年龄因素两组匹配,因此模型构建中未考虑该因素。未来的中国女性乳腺癌风险预测模型可以考虑在Zheng模型基础上加上年龄因素。
获得人群综合风险得分的分布之后,需综合现有的社会资源,确定合适的综合风险分层方法。如将人群综合风险>75%的人群定义为高危人群。或经过大量的数据模拟之后确定理论的高危风险界值。
7. 基于不同风险分层方法,评价目标SNP对乳腺癌覆盖率的影响。在乳腺癌总体筛查效果的评价指标上,虽然乳腺癌检出率是一个最直接的传统评价指标,但是如果单纯追求高检出率就很可能导致研究者片面地关注极端高危人群,而遗漏大部分其他乳腺癌患者。因为既往研究提示:不论是乳腺癌患者还是健康人群,GRS的人群分布以及综合风险分布,均呈现正态分布[18, 20]。极端高危的人群只占人群风险分布很小一部分,绝大部分人群或乳腺癌患者均处于风险中位。而筛查最直接的目的仍是为了捕获更多的乳腺癌患者。因此,在乳腺癌筛查效果的评价上,除了关注乳腺癌检出率等传统评价指标外,乳腺癌覆盖率(筛查发现的乳腺癌患者例数占所有潜在乳腺癌患者的比例)是一个非常重要的筛查效果补充指标[16]。
初步评价乳腺癌覆盖率的方法包括分别构建单纯环境危险因素的风险分层方法(模型1)及环境危险因素+GRS的综合风险分层方法(模型2)。假定将人群风险>75%的人群定义为高危人群,并筛查,可分别计算两种风险分层方法下所覆盖的乳腺癌患者比例,而二者之差,即为筛查中引入GRS后,乳腺癌覆盖率改善的理论估计。
综上所述,随着GWAS发现乳腺癌易感性相关SNP位点的增加,探讨这些SNP的公共卫生学意义,尤其是应用于人群筛查的价值,逐渐成为后GWAS时代的热点问题之一。而乳腺癌作为最具有筛查价值的肿瘤,探讨GWAS发现SNP在人群乳腺癌筛查中的价值显得尤为重要。虽然目前国内外存在一些相关研究,但仍然有许多问题有待进一步阐明。本文在结合国内外相关研究进展基础上,提出了如何结合PVi,以及新的人群风险分层改善评价指标(包括IDI和NRI),选取适合用于乳腺癌筛查的合适数量SNP位点的方法。以及如何评价GWAS来源SNP用于人群筛查时,对乳腺癌覆盖率影响的初步估算方法。未来需要更多的研究以验证该方法的合理性。同时也需要更多的研究来证实不同遗传风险的女性及不同筛查方法的准确性是否存在差异,从 而进一步拓展GWAS来源SNP的应用价值。
| [1] Ferlay J,Shin HR,Bray F,et al. Estimates of worldwide burden of cancer in 2008:GLOBOCAN 2008[J]. Int J Cancer,2010,127(12):2893-2917. |
| [2] Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening:an independent review[J]. Lancet,2012,380(9855):1778-1786. |
| [3] Pharoah PD, Sewell B,Fitzsimmons D,et al. Cost effectiveness of the NHS breast screening programme:life table model[J]. BMJ,2013,346:f2618. |
| [4] Wong IOL, Kuntz KM, Cowling BJ,et al. Cost effectiveness of mammography screening for Chinese women[J]. Cancer,2007,110(4):885-895. |
| [5] Cai QY,Zhang B,Sung H,et al. Genome-wide association analysis in East Asians identifies breast cancer susceptibility loci at 1q32.1,5q14.3 and 15q26.1[J]. Nat Genet,2014,46(8):886-890. |
| [6] Long JR,Cai QY,Sung H,et al. Genome-wide association study in east Asians identifies novel susceptibility loci for breast cancer[J]. PLoS Genet,2012,8(2):e1002532. |
| [7] Cai QY,Long JR,Lu W,et al. Genome-wide association study identifies breast cancer risk variant at 10q21.2:results from the Asia Breast Cancer Consortium[J]. Hum Mol Genet,2011,20(24):4991-4999. |
| [8] Long JR, Cai QY, Shu XO,et al. Identification of a functional genetic variant at 16q12.1 for breast cancer risk:results from the Asia Breast Cancer Consortium[J]. PLoS Genet,2010,6(6):e1001002. |
| [9] Zheng W, Long JR, Gao YT, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1[J]. Nat Genet,2009,41(3):324-328. |
| [10] Michailidou K, Hall P, Gonzalez-Neira A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk[J]. Nat Genet,2013,45(4):353-361. |
| [11] Ahmed S, Thomas G, Ghoussaini M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2[J]. Nat Genet,2009,41(5):585-590. |
| [12] Stacey SN, Manolescu A, Sulem P,et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer[J]. Nat Genet,2008,40(6):703-706. |
| [13] Hunter DJ,Kraft P,Jacobs KB,et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer[J]. Nat Genet,2007,39(7):870-874. |
| [14] Ghoussaini M,Fletcher O,Michailidou K,et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci[J]. Nat Genet,2012,44(3):312-318. |
| [15] Zheng W,Zhang B,Cai QY,et al. Common genetic determinants of breast-cancer risk in East Asian women:a collaborative study of 23 637 breast cancer cases and 25 579 controls[J]. Hum Mol Genet,2013,22(12):2539-2550. |
| [16] Garcia-Closas M,Gunsoy NB,Chatterjee N. Combined associations of genetic and environmental risk factors:implications for prevention of breast cancer[J]. J Natl Cancer Inst,2014,106(11):dju305. |
| [17] Wacholder S,Hartge P,Prentice R,et al. Performance of common genetic variants in breast-cancer risk models[J]. N Engl J Med,2010,362(11):986-993. |
| [18] Mealiffe ME, Stokowski RP, Rhees BK, et al. Assessment of clinical validity of a breast cancer risk model combining genetic and clinical information[J]. J Natl Cancer Inst,2010,102(21):1618-1627. |
| [19] Gail MH. Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model[J]. J Natl Cancer Inst,2009,101(13):959-963. |
| [20] Pharoah PDP,Antoniou AC,Easton DF,et al. Polygenes,risk prediction,and targeted prevention of breast cancer[J]. N Engl J Med,2008,358(26):2796-2803. |
| [21] Gail MH. Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk[J]. J Natl Cancer Inst,2008,100(14):1037-1041. |
| [22] Zheng W,Wen WQ,Gao YT,et al. Genetic and clinical predictors for breast cancer risk assessment and stratification among Chinese women[J]. J Natl Cancer Inst,2010,102(13):972-981. |
| [23] Pencina MJ,D'Agostino RB,D'Agostino RB,et al. Evaluating the added predictive ability of a new marker:from area under the ROC curve to reclassification and beyond[J]. Stat Med,2008,27(2):157-172. |
| [24] Kennedy KF,Pencina MJ. SDA-07 A SAS® macro to compute added predictive ability of new markers predicting a dichotomous outcome[S/OL].[2015-03-18]. http://analytics.ncsu.edu/sesug/2010/SDA07.Kennedy.pdf. |