 2015, Vol. 36
2015, Vol. 36文章信息
- 李娇元, 常江, 朱颖, 杨洋, 龚雅洁, 柯俊涛, 娄娇, 钟荣, 龚静, 夏肖萍, 缪小平.
- Li Jiaoyuan, Chang Jiang, Zhu Ying, Yang Yang, Gong Yajie, Ke Juntao, Lou Jiao, Zhong Rong, Gong Jing, Xia Xiaoping, Miao Xiaoping.
- 基于常见遗传变异和传统风险因素的中国南方汉族人群结直肠癌风险预测模型研究
- Risk prediction of colorectal cancer with common genetic variants and conventional non-genetic factors in a Chinese Han population
- 中华流行病学杂志, 2015, 36(10): 1053-1057
- Chinese Journal of Epidemiology, 2015, 36(10): 1053-1057
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2015.10.003
-
文章历史
- 投稿日期: 2015-06-15




2. 环境与健康教育部重点实验室;
3. 浙江大学医学院附属第四医院检验科
2 Key Laboratory of Environment and Health, Ministry of Education and Ministry of Environmental Protection, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China;
3 Clinical Laboratory of the Fourth Affiliated Hospital, Zhejiang University School of Medicine
结直肠癌是世界范围内常见的消化系统恶性肿瘤[1]。近年来我国结直肠癌的发病率和死亡率均呈平稳上升趋势[2]。有效的结直肠癌风险预测模型是用于结直肠癌高危人群筛查,实现早发现、早预防和早治疗的有效手段。已有的结直肠癌预测模型主要探讨环境因素在风险预测中的作用[3, 4]。而结直肠癌的发生是多种环境和遗传因素共同作用的结果,后者在肿瘤形成和发展中也扮演着重要角色[5]。在全基因组关联研究(GWAS)成功应用于肿瘤遗传研究的背景下,GWAS发现的遗传易感位点赋予了风险预测模型新的含义。为此本研究旨在利用GWAS发现的中国南方汉族人群结直肠癌易感位点,联合性别、年龄、吸烟、饮酒等常见风险因素建立结直肠癌风险预测模型,为构建适合中国人群的结直肠癌早期预警预测体系提供理论参考依据。 对象与方法
1. 研究对象:共纳入4 946人,其中结直肠癌1 066例(病例组),正常对照3 880例(对照组)。分别来自2009年1月至2012年12月在华中科技大学同济医院、协和医院确诊的结直肠癌病例和同期随机抽取协和医院的健康体检人群。病例组纳入标准为汉族,并经组织病理学确诊的结直肠癌患者,排除非原发性结直肠癌及其他恶性肿瘤者。对照组均通过基本体检确认无肿瘤病史和症状。研究对象的人口学资料(包括性别、年龄、吸烟和饮酒状态)来自病历或体检报告。从未吸或平均每天吸<1支且吸烟时间<1年者定义为非吸烟者,否则为吸烟者;一生中每周饮酒>2次且持续>1年为饮酒者,否则为非饮酒者。研究对象在参加该项目前均签署知情同意书,该项目经华中科技大学伦理委员会批准。
2. 遗传变异位点的选择与基因分型:通过查阅NHGRI GWAS数据库中相关文献[6],去除连锁不平衡的位点,选择了21个在东亚人群中与结直肠癌相关的常见单核苷酸多态性位点(SNP)作为风险预测模型的候选位点[7, 8, 9]。在TaqMan基因分型系统(ABI 7900HT Real Time PCR System,Applied Biosystems)完成基因分型。PCR反应为5 μl体系,反应条件为95 ℃预变性10 min;95℃ 15 s和60℃ 1 min,共40个循环。由于21个候选位点中2个位点(rs10774214 和rs1665650)基因分型失败,最终19个位点纳入后续研究。排除基因分型成功率较低(<90%)的研究对象,共1 065例结直肠癌患者和3 873名对照纳入研究。
3. 遗传位点与结直肠癌易感性的关联分析:运用logistic回归中的加性模型(additive model)计算19个位点与结直肠癌易感性之间的关联。模型中校正了年龄、性别、吸烟及饮酒状态,并以P<0.05为差异有统计学意义的标准筛选纳入风险模型的位点。在19个遗传位点中,7个位点与结直肠癌具有显著关联,因而列入风险模型。
4. 结直肠癌风险预测模型的构建:本研究采用遗传风险评分(genetic risk score,GRS)计算7个显著位点的联合作用。该方法是将每个SNP位点看作独立的结直肠癌危险因素,利用0、1、2三个线性数值分别代表某个体携带某一SNP风险等位基因的个数。而位点联合作用的计算方法包括GRS和权重遗传风险评分(weighted genetic risk score,wGRS)[10]。GRS假设每个位点对结直肠癌易感性有相同贡献,个体的GRS即为其携带风险等位基因个数之和。wGRS假设每个位点对结直肠癌易感性的影响不同,但与其OR值有关,因此wGRS为每个SNP在logistic回归中β值加权后的平均风险等位基因个数。在此基础上,为了避免个别缺失值的影响以及便于比较,本文将wGRS分别除以 β 值之和的2倍并乘以SNP等位基因个数。GRS和wGRS均根据其在研究对象中的分布进行了四分位划分,分别命名为QGRS和QwGRS。所有风险评分与结直肠 癌易感性的关联均在调整了性别、年龄、吸烟、饮酒状态的情况下由非条件logistic回归计算得出。
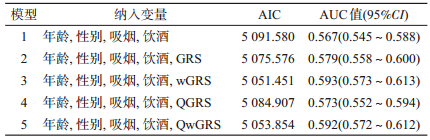
本研究利用遗传因素(7个SNP位点)和传统风险因素(性别、年龄、吸烟和饮酒)分别构建了5种风险模型。模型1仅纳入性别、年龄、吸烟和饮酒因素;模型2在纳入以上因素的基础上加入GRS;模型3在纳入传统风险因素的基础上加入wGRS;模型4在纳入传统风险因素的基础上加入QGRS;模型5在纳入传统风险因素的基础上加入QwGRS。并采用赤池信息准则(Akaike’s Information Criterion,AIC)作为评价以上模型拟合优良性的指标。利用受试者工作特征(ROC)曲线及计算曲线下面积(AUC)评价模型的预测效果。
5. 统计学方法:采用PLINK软件完成候选位点的关联分析和GRS[11];风险模型构建在SAS 9.1软件中完成;利用SPSS 13.0软件完成ROC及AUC计算。 结果
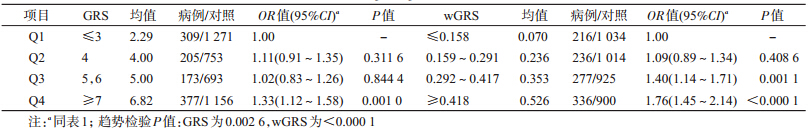
1. 遗传位点与结直肠癌易感性的关联:以P<0.05为初筛标准,19个SNP位点中7个(rs647161、rs10505477、rs6983267、rs10795668、rs7229639、rs4939827、rs2423279)与结直肠癌易感性存在显著关联(表 1)。其中最显著的位点为20号染色体上的rs2423279(OR=1.24,95%CI:1.12~1.37,P=2.77×10-5)。为了进一步分析显著位点的联合作用,计算了7个位点的GRS和wGRS。显示病例组和对照组的GRS分布略有不同(图 1),相比对照组,病例组的GRS分布偏向右移;随着GRS的升高,病例组所占的相对比例也逐渐升高。对GRS和wGRS根据四分位进行分组,并计算各组人群结直肠癌的患病风险。结果显示,随着GRS或wGRS的增加,人群罹患结直肠癌的风险也逐渐增加(表 2)。以GRS≤3的人群为参照,评分 4~5、6和≥7的人群罹患结直肠癌的风险分别为OR=1.11(95%CI:0.95~1.35),P=0.311 6;OR=1.02(95%CI:0.83~1.26),P=0.844 4和OR=1.33(95%CI:1.12~1.58),P= 0.001 0;趋势检验:P=0.002 6。以wGRS<0.158的人群为参照,wGRS为0.159~0.291的人群患结直肠癌风险的OR=1.09(95%CI:0.89~1.34),P=0.408 6;wGRS为0.292~0.417的人群OR=1.40(95%CI:1.14~1.71),P=0.001 1;wGRS≥0.418的人群OR=1.76(95%CI:1.45~2.14),P<0.000 1;趋势检验:P<0.000 1。

 |
| 注:Ⓐ病例组和对照组 GRS分布;Ⓑ不同 GRS下病例组和对照组所占比例 图 1 结直肠癌显著易感位点的遗传风险评分在病例 和对照中的分布 |
2. 风险预测模型构建及模型间的比较:比较5种不同方式建模的AIC值,选出拟合优度最优者(表 3)。以仅纳入性别、年龄、吸烟、饮酒4种传统风险因素模型的AIC值为参考(AIC=5 091.580),表明以不同方式联合环境因素和遗传因素构建的模型均优于单独纳入传统风险因素的模型,其中以传统风险因素联合wGRS的模型为拟合度最好模型,AIC=5 051.451。以不同方式建模的ROC曲线见图 2,相比于仅纳入传统风险因素(模型1),在这些风险因素基础上加入GRS的模型(模型2),其AUC提升了1.2% (0.579 vs. 0.567)。而联合传统风险因素和wGRS的模型AUC进一步提升至0.593(95%CI:0.573~0.613)。综合以上结果,传统风险因素联合wGRS进入预测模型具有最优的拟合性和最好的预测效果,因此应以此方式建立结直肠癌的风险预测模型。
 |
| 图 2 不同结直肠癌风险预测模型的 ROC曲线 |
本文采用病例对照研究分析了经GWAS发现与结直肠癌易感性关联的21个亚洲人群阳性位点,显示7个SNP位点与中国南方汉族人群结直肠癌显著相关。为此利用这7个位点计算个体GRS,并联合传统风险因素以5种不同方式构建了结直肠癌风险预测模型。结果表明,基于易感位点计算得出的GRS与结直肠癌易感性显著相关。传统风险因素联合遗传因素的预测模型优于传统环境因素模型。其中性别、年龄、吸烟、饮酒4个常见风险因素和wGRS组成最优的结直肠癌风险预测模型。
目前基于GWAS的风险预测模型已经应用于2型糖尿病、前列腺癌和乳腺癌等复杂性疾病的风险预测[12, 13, 14]。本研究基于GWAS的研究结果构建了中国南方汉族人群结直肠癌的风险预测模型。值得注意的是,本研究中19个GWAS发现的易感位点只有7个验证出了阳性关联。可能的原因是样本人群存在的异质性和病例组样本量的局限所致。遗传因素联合传统风险因素构建的结直肠癌风险模型优于传统风险因素模型是本研究的主要发现之一。然而,即使是最优模型(由性别、年龄、吸烟、饮酒和wGRS组成),相比于传统风险因素模型,其AUC仅仅提高了约3%。该结果与乳腺癌的预测模型极其相似[14, 15]。原因可能是GWAS发现的易感位点均为常见变异,对疾病易感性的效应亦极其微弱(大部分GWAS发现的肿瘤易感位点的OR值均<2.0,仅能解释一小部分遗传易感性)[16]。本研究中,与wGRS四分位分组中最低的一组相比,wGRS值最高的一组仅提升了约76%的结直肠癌风险,说明少数几个易感位点间的联合作用远远不足以代表全部的遗传风险,更多的结直肠癌易感位点还有待进一步探讨。
本研究存在不足。病例对照研究在收集人口学及环境暴露资料时可能存在信息偏倚。影响结直肠癌的环境因素多种多样[17, 18, 19, 20],而本研究只探讨了性别、年龄、吸烟及饮酒4种常见因素。由于GWAS的局限性,纳入的遗传因素也非常有限。此外,本研究人群为南方汉族人群,其存在的人群异质性使得结果的外推受到一定限制。因此在大规模多中心的前瞻性队列研究中,构建多种环境因素和遗传因素组成的风险预测模型将是今后的研究方向。
| [1] Bray F,Ren JS,Masuyer E,et al. Global estimates of cancer prevalence for 27 sites in the adult population in 2008[J]. Int J Cancer,2013,132(5):1133-1145. |
| [2] Dong J,Hu ZB,Wu C,et al. Association analyses identify multiple new lung cancer susceptibility loci and their interactions with smoking in the Chinese population[J]. Nat Genet,2012,44(8): 895-899. |
| [3] Lan Q,Hsiung CA,Matsuo K,et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia[J]. Nat Genet,2012,44(12): 1330-1335. |
| [4] Dong J,Jin GF,Wu C,et al. Genome-wide association study identifies a novel susceptibility locus at 12q23.1 for lung squamous cell carcinoma in han chinese[J]. PLoS Genet,2013,9 (1):e1003190. |
| [5] Hu ZB,Xia YK,Guo XJ,et al. A genome-wide association study in Chinese men identifies three risk loci for non-obstructive azoospermia[J]. Nat Genet,2012,44(2):183-186. |
| [6] Broderick P,Wang YF,Vijayakrishnan J,et al. Deciphering the impact of common genetic variation on lung cancer risk:a genome-wide association study[J]. Cancer Res,2009,69(16): 6633-6641. |
| [7] Shiraishi K,Kunitoh H,Daigo Y,et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population[J]. Nat Genet,2012, 44(8):900-903. |
| [8] Yoon KA,Park JH,Han J,et al. A genome-wide association study reveals susceptibility variants for non-small cell lung cancer in the Korean population[J]. Hum Mol Genet,2010,19(24): 4948-4954. |
| [9] Miki D,Kubo M,Takahashi A,et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations[J]. Nat Genet,2010,42(10):893-896. |
| [10] Wang YP,Lu Y,Zhang Y,et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation[J]. Nat Genet,2015,47(6): 625-631. |
| [11] Landi MT,Chatterjee N,Yu K,et al. A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma[J]. Am J Hum Genet, 2009,85(5):679-691. |
| [12] Deng QF,Guo H,Dai JC,et al. Imputation-based association analyses identify new lung cancer susceptibility variants in CDK6 and SH3RF1 and their interactions with smoking in Chinese populations[J]. Carcinogenesis, 2013, 34 (9) : 2010-2016. |
| [13] Hu ZB,Wu C,Shi YY,et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese[J]. Nat Genet,2011,43(8):792-796. |
| [14] Delaneau O,Marchini J. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel[J]. Nat Commun,2014,5:3934. |
| [15] Howie BN,Donnelly P,Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies[J]. PLoS Genet,2009,5(6):e1000529. |
| [16] 1 000 Genomes Project Consortium,Abecasis GR,Auton A,et al. An integrated map of genetic variation from 1 092 human genomes[J]. Nature,2012,491(7422):56-65. |
| [17] Welter D,MacArthur J,Morales J,et al. The NHGRI GWAS Catalog,a curated resource of SNP-trait associations[J]. Nucleic Acids Res,2014,42(Database issue):D1001-1006. |
| [18] Dai J,Hu Z,Jiang Y,et al. Breast cancer risk assessment with five independent genetic variants and two risk factors in Chinese women[J]. Breast Cancer Res,2012,14(1):R17. |
| [19] Bach PB,Kattan MW,Thornquist MD,et al. Variations in lung cancer risk among smokers[J]. J Natl Cancer Inst,2003,95(6): 470-478. |
| [20] Cassidy A,Myles JP,van Tongeren M,et al. The LLP risk model: an individual risk prediction model for lung cancer[J]. Br J Cancer,2008,98(2):270-276. |
| [21] Spitz MR,Hong WK,Amos CI,et al. A risk model for prediction of lung cancer[J]. J Natl Cancer Inst,2007,99(9):715-726. |
| [22] Hoggart C,Brennan P,Tjonneland A,et al. A risk model for lung cancer incidence[J]. Cancer Prev Res (Phila),2012,5(6): 834-846. |