 2015, Vol. 36
2015, Vol. 36文章信息
- 马江涛, 袁芳, 陈慧, 马学旻, 詹军, 李丽. 2014.
- Ma Jiangtao, Yuan Fang, Chen Hui, Ma Xuemin, Zhan Jun, Li Li. 2014.
- 宁夏回族自治区2013年手足口病患者柯萨奇病毒A组10型分离株VP1区基因特征分析
- Genetic characteristics of VP1 region of coxsackievirus A10 strains isolated from hand foot and mouth disease patients in Ningxia Hui Autonomous Region, 2013
- 中华流行病学杂志, 2015, 36(7): 734-737
- Chinese Journal of Epidemiology, 2015, 36(7): 734-737
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2015.07.015
-
文章历史
- 投稿日期:2014-12-03




手足口病(HFMD)是由人类肠道病毒引起的一种常见传染病,主要病原是肠道病毒71 型(EV71)和柯萨奇病毒A组16 型(Cox A16)。但近年来,该病病原谱逐渐发生了变化,非EV71 和非Cox A16 的其他肠道病毒比例逐渐增加[1, 2],而且导致重症或死亡病例发生[3]。2013 年宁夏回族自治区(宁夏)共报告HFMD病例6 069 例,实验室诊断病例796 例,其中104 例EV71 型,412 例Cox A16,其他肠道病毒280例。为了解肠道病毒型别构成情况和常见病原体的基因特征,本研究对2013 年分离到的柯萨奇病毒A组10 型(Cox A10)毒株进行了VP1 区基因测序和特征分析。
材料与方法1. 标本来源:2013 年宁夏各市(县)共采集HFMD 临床病例标本1 155 份,其中粪便标本272份、咽拭子883 份。
2. 病毒核酸提取和鉴定:标本按照国家《手足口病防治指南》(2009 版)处理后,吸取200 μl 上清液提取病毒RNA,采用硕世生物科技公司生产的EV71、Cox A16 和肠道病毒通用检测试剂进行核酸检测,步骤及条件参见试剂说明书。
3. 病毒分离:取200 μl 核酸检测阳性标本上清液,接种至长满单层、形态良好的RD细胞,置36 ℃培养箱内,连续7 d 观察细胞病变(CPE),当CPE≥75%时收获病毒,并置于-20 ℃保存;若连续传2 代仍无CPE出现,则判为阴性。
4. RT-PCR检测:病毒分离阳性分离物反复冻融后,提取病毒RNA,根据参考文献,使用兼并引物进行肠道病毒VP1 区RT-PCR 扩增[4]。扩增试剂为北京全式金生物技术有限公司一步法RT-PCR 试剂盒,反应条件:42 ℃ 45 min,95 ℃ 2 min;94 ℃ 30 s,50 ℃ 30 s,72 ℃ 80 s,40 个循环;72 ℃ 延伸10 min。PCR产物用1.5%琼脂糖凝胶电泳分析结果。
5. 序列测定与分析:电泳阳性PCR产物送生工生物工程(上海)股份有限公司进行核苷酸序列测定。测序结果利用SeqMan 软件进行拼接,拼接后的序列进行BLAST 同源性检索。对鉴定为CoxA10 的宁夏株与各亚型代表株利用MegAlign 软件进行核苷酸和氨基酸同源性分析,利用Mega 6.0 软件采用邻接法构建系统发生树,建树的可靠性通过1 000 bootstrap 来评估。用于同源性分析和构建系统发生树的参比序列均来自GenBank。
结果1. HFMD病原构成:2013 年共采集临床HFMD病例标本1 155 份,经real-time PCR 检测,阳性标本为796 份,阳性率为68.92%;其中EV71、Cox A16 和其他肠道病毒分别为104、412 和280 份,所占比例分别为13.06%、51.76%和35.18%。
2. 其他肠道病毒的分离和鉴定:使用RD 细胞从280 份标本中共分离到其他肠道病毒28 株,通过对其VP1 区基因测序和BLAST分析,共鉴定出4 个基因型别,其中6 株(21.43% )Cox A10,4 株(14.29% )Cox A4,5 株(17.86% )Cox A6,2 株(7.14%)Cox A14,未分型肠道病毒11 株(39.29%)。
3. Cox A10 宁夏株VP1 区核苷酸、氨基酸序列分析:对6 株Cox A10 宁夏株及从GenBank 中检索到的Cox A10 原型株(Kowalik/USA/1950)和其他地区流行株进行VP1 区核苷酸和氨基酸序列同源性分析[3]。发现6 株Cox A10 宁夏株间具有较高的同源性,相互间核苷酸同源性为97.0%~99.8%,氨基酸同源性为99.0%~99.7%。与A、B、C、D各型别代表株的同源性比较发现,6 株Cox A10 宁夏株与C型代表株的核苷酸和氨基酸同源性最高,分别为94.4%~98.9%和98.0%~99.7%(表 1)。
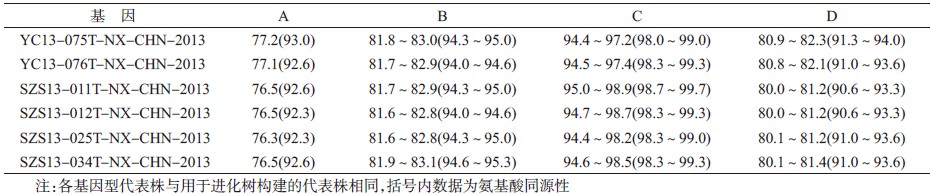
4. Cox A10 宁夏株亲缘进化分析:VP1 区基因系统发生树分析显示,Cox A10 被划分为4 个基因型(A~D),我国目前分离到的Cox A10 毒株均属于B基因型和C基因型,其中B基因型主要包括山东省2004-2008 年分离株,C基因型主要包括山东省和江苏省2008-2012 年分离株和河北省2013 年分离株,A基因型仅有美国一株Kowalik 原型株,与其他型别同源性差别较大,D基因型主要包括国外一些国家分离到的毒株,如西班牙2008 年分离株、法国2010 年分离株和俄罗斯2010 年分离株。从基因进化关系来看,2009 年以后我国流行的Cox A10 主要以C基因型为主。本次分离到的6 株宁夏株与C型代表株处于同一分支,属于C型Cox A10,与2009 年以后我国其他地区流行型别一致[3, 5]。6 株宁夏株之间,银川地区分离株(YC13-075T和YC13-075T3)与石嘴山地区分离株(SZS13-012T、SZS13-025T 和SZS13-025T)同源差距较大,具有明显的地域性,形成了不同支系,表明有多条传播链在宁夏地区共循环。
讨论除了EV71 和Cox A16 之外,近年来Cox A10、Cox A6 和Cox B3 等病毒也逐渐演变成为HFMD的常见病原体[1, 6, 7, 8, 9]。新加坡学者在2009 年提出[10],Cox A10 是除EV71 外可单独引起重症病例的病原体,在疾病防控中,与EV71 和Cox A16 地位同等重要。
本次研究使用兼并引物对宁夏非EV71 和非Cox A16肠道病毒进行了RT-PCR 鉴定,结果显示,在宁夏HFMD病例的病原中,Cox A10 构成了除EV71 和Cox A16 之外的最主要的病原体,与我国河北、广东、山东等省份监测结果一致[3, 5, 11, 12]。针对2013 年我国各省份HFMD 病原中其他肠道病毒所占比例大幅度增高的现象,希望引起有关部门的重视。
通过对病毒基因进化和同源性分析发现,Cox A10 病毒划分为4 个基因型(A~D),与Tian 等[3]研究结果一致。我国流行Cox A10 具有明显的时间进化特征,2004-2008 年主要以B 基因型流行为主,2009年以后主要以C 基因型为主。根据不同年度毒株进化关系来看,相比较2008 - 2010 年山东、江苏等地流行的Cox A10,宁夏流行株与2012 年河北省石家庄市流行株具有更高的同源性,说明宁夏地区流行的Cox A10 与国内同一时期流行Cox A10 一致。但通过病毒系统进化分析发现,宁夏株进化又相对独立,与同源性最高的石家庄株相比,核苷酸同源性为97.0%~98.9%,氨基酸同源性为99.0%~99.7%,形成了较为独立的进化分支,说明可能宁夏地区性流行时间较长,为更加清楚地阐述其在宁夏地区的流行情况,需要做好连续性监测。
6 株宁夏株之间也具有明显的地域特征,2 株银川分离株与4 株石嘴山分离株形成了两支明显的传播链Lineage 1 和Lineage 2(图 1)。同一传播链毒株之间核苷酸同源性较高,Lineage 1 毒株间的同源性为99.3% ~99.8% ,Lineage 2 毒株间的同源性为99.3% ;不同传播链毒株之间同源性也有差异,Lineage 1 和Lineage 2 毒株间的同源性为97.0%~97.3%。而且不同地区之间毒株形成了两支明显的传播链。表明宁夏流行Cox A10 具有明显的地域性,存在至少两条传播链发生共循环。
 |
| 图 1 6株宁夏Cox A10分离株与各型代表株的VP1基因(894nt)系统发生树 |
引起HFMD的病原体种类繁多,其主导病原体在不断地发生着变化。随着主要引起HFMD病原体——EV71 和Cox A16 的广泛流行,其通过自然感染而建立的免疫屏障达到一定的保护水平后,另外一种病原体的危害性会凸显出来。因此,加强不同时间、不同地区其他肠道病毒的监测,全面了解其基因进化特征和遗传信息,对于当地HFMD的防控具有重要的意义。
| [1] Lu QB,Zhang XA,Wo Y,et al. Circulation of CoxsackievirusA10 and A6 in hand-foot-mouth disease in China,2009-2011[J]. PLoS One,2012,7(12):e52073. |
| [2] Hu YF,Yang F,Du J,et al. Coxsackievirus B5,associated withneurological hand,foot and mouth disease,China[J]. J Infect,2012,65(2):189-191. |
| [3] Tian HF,Zhang Y,Sun Q,et al. Prevalence of multipleenteroviruses associated with hand-foot-mouth disease inShijiazhuang city,Hebei province in China:Outbreaks ofCoxsackieviruses A10 and B3[J]. PLoS One,2014,9(1):e84233. |
| [4] Oberste MS,Maher K,Williams AJ,et al. Species-specificRT-PCR amplification of human enteroviruses:a tool for rapidspecies identification of uncharacterized enteroviruses[J]. J GenVirol,2006,87(1):119-128. |
| [5] He YQ,Chen L,Xu WB,et al. Emergence,circulation,andspatiotemporal phylogenetic analysis of Coxsackievirus A6- andCoxsackievirus A10-associated hand,foot,and mouth diseaseinfections from 2008 to 2012 in Shenzhen,China[J]. J ClinMicrobio,2013,51(11):3560-3566. |
| [6] Blomqvist S,Klemola P,Kaijalainen S,et al. Co-circulation ofCoxsackieviruses A6 and A10 in hand,foot and mouth diseaseoutbreak in Finland[J]. J Clin Virol,2010,48(1):49-54. |
| [7] Chen C,Xie HP,Cui M,et al. An investigation on a case of handfoot-mouth disease caused by Coxsackie-virus A6 associatedwith a vaccine-derived poliovirus co-infection [J]. Chin JEpidemiol,2014,35(1):61-65.(in Chinese)陈纯,谢华萍,崔敏,等. 柯萨奇A6 型手足口病耦合脊髓灰质炎疫苗衍生株感染一例调查[J]. 中华流行病学杂志,2014,35(1):61-65. |
| [8] Mirand A,Henquell C,Archimbaud C,et al. Outbreak of hand,foot and mouth disease herpangina associated withCoxsackievirus A6 and A10 infections in 2010,France:a largecitywide,prospective observational study[J]. Clin MicrobiolInfect,2012,18(5):E110-118. |
| [9] Hu YF,Yang F,Du J,et al. Complete genome analysis ofCoxsackievirus A2,A4,A5,and A10 strains isolated from hand,foot and mouth disease patients in China revealing frequentrecombination of human enterovirus A[J]. J Clin Microbiol,2011,49(7):2426-2434. |
| [10] Anq LW,Koh BK,Chan KP,et al. Epidemiology and control ofhand,foot and mouth disease in Singapore,2001-2007[J]. AnnAcad Med Singapore,2009,38(2):106-112. |
| [11] Yang H,Tao ZX,Wang HY,et al. The genetic characterization ofVP1 region of Coxsackie virus A10 isolated from hand,foot andmouth disease cases in Shandong province of China[J]. Chin JInfect Dis,2010,28(7):385-389.(in Chinese)杨赫,陶泽新,王海岩,等. 手足口病患儿柯萨奇病毒A10 型山东地方株VP1 区基因特征分析[J]. 中华传染病杂志,2010,28(7):385-389. |
| [12] Zhen RN,Zhang Y,Xie HP,et al. Sequence analysis of VP1region of Coxsackievirus A4 and Coxsackievirus A10 inGuangzhou city,2010-2012[J]. Chin J Prev Med,2014,48(6):445-450.(in Chinese)甄若楠,张颖,谢华萍,等. 2010-2012 年广州市柯萨奇病毒A4、A10 型VP1 基因特征分析[J]. 中华预防医学杂志,2014,48(6):445-450. |