 2024, Vol. 35
2024, Vol. 35 


b Key Laboratory of Beibu Gulf Environment Change and Resources Utilization (Nanning Normal University), Ministry of Education, College of Chemistry and Life Science, Nanning Normal University, Nanning 530000, China
The environmental problems caused by massive CO2 emissions have forced the implementation of "carbon neutrality" [1], and with the call of the goal of "carbon neutrality", the resourceful use of CO2 (e.g., conversion into higher value-added fuels, vital industrial feedstock or other chemicals) has become an area of great significance in sustainable development strategies [2, 3]. As an indispensable nitrogen fertiliser, urea is essential to meet the needs of a growing population. However, the traditional industrial synthesis of urea requires extremely harsh conditions (N2 + H2 → NH3, 150–350 bar, 350–550 ℃, followed by NH3 + CO2 → CO(NH2)2, 150–250 bar, 150–200 ℃), which is contrary to the aim of energy saving and emission reduction. Therefore, a new method of urea synthesis by electrocatalytic C—N coupling has emerged, and the employment of CO2 as the carbon source for the formation of C—N bond is undoubtedly the best choice [4]. Recently, D.B. Kayan et al. realized the co-reduction of N2 and CO2 at ultra-low potentials of −0.165 V using the polyaniline (PAni) and polypyrrole (PPy) coated platinum electrodes, and the main products obtained were urea, ammonia and formic acid [5]; Wang et al. used PdCu/TiO2 as an electrocatalyst to directly couple CO2 and N2 to produce urea under environmental conditions [6], which achieved a high urea formation rate of 3.36 mmol g−1 h−1 and a considerable FE of 8.92%; Zhang et al. successfully achieved high-efficiency synthesis of urea (the formation rate and FE were 4.94 mmol h−1 g−1 and 17.18%, respectively) through the co-activated reduction of N2 and CO2 on BiFeO3/BiVO4 p-n heterojunction electrocatalysts [7]. Considering that the wastewater discharged by many industries often contains a large amount of NO3−/NO2−, if they are used as the nitrogen source for the preparation of urea by C—N coupling reaction, this could theoretically not only reduce the cost of raw materials, but also incidentally solve environmental problems such as eutrophication in water bodies. In such circumstances, the NO3−/NO2− reduction reaction (NO3−/NO2− RR) immediately attracted the attention of researchers [8]. As early as decades ago, M. Shibata et al. had discovered that the product of C—N coupling by co-reduction of CO2 and NO3− or NO2− was urea [9-12]. And lately, Yu and co-workers attained urea production by electrochemically coupling nitrate with carbon dioxide on In(OH)3, high average yield (533.1 µg h−1 mg−1), high FE (53.4%) and high selectivity (C/N-selectivity of ~100%/82.9%) were achieved at −0.6 V vs. RHE [13]; Zhang and co-workers prepared a self-supported oxygen vacancy-rich zinc oxide (ZnO-V) porous nanosheets as high performance electrocatalyst for the synthesis of urea from CO2 and NO2−, which achieved a FE of 23.26% at −0.79 V vs. RHE [14]; Geng and co-workers successfully converted CO2 and NO3− to urea using a symbiotic graphitic carbon encapsulated amorphous iron and iron oxide nanoparticles on carbon nanotubes (Fe(a)@C-Fe3O4/CNTs) with a FE of 16.5% ± 6.1%, and urea yield of 1341.3 ± 112.6 µg h−1 mgcat−1 [15]. Nevertheless, the successful application of the available research results to industrial production requires additional efforts due to several reasons: (1) The rate of urea yield and selectivity need to be further improved; (2) The seeking of active sites for C—N coupling reactions and the suppression of competitive hydrogen evolution reaction (HER) remains a challenge; (3) The mechanism of electrocatalytic production of urea is not yet clear, which makes it difficult to theoretically guide the synthesis of high-efficiency catalysts [4, 13].
Compared to currently available methods in industry, the electrocatalytic co-reduction of CO2 and NO3− for the synthesis of urea has economic and environmental advantages and is more in line with the requirements of sustainable development: (1) Wide source of raw materials (CO2-fossil fuel combustion, NO3−- industrial waste water discharge). It is a win-win strategy for both the environment and the economy by turning waste into treasure and reducing environmental pressure simultaneously; (2) The reaction requires no high temperature or pressure and can be completed at room temperature, which is an energy-saving strategy. The Haber-Bosch process consumes 2% of the world's energy, and approximately 80% of the ammonia synthesized by this process is used in the synthesis of urea.
Hereby, the graphene-In2O3 electrocatalysts were meticulously designed and synthesized for the co-activation reduction of CO2 and NO3− to generate urea with high efficiency. On the one hand, graphene acts as a support and has an excellent dispersion effect to the In2O3 nanoparticles, allowing more exposure of the active sites, which maximizes the intrinsic activity of In2O3. On the other hand, experiments and theoretical calculations together reveal that the key intermediates in the C—N coupling reaction here are most likely to be *NH2 and *CO2, between which the coupling reaction has a rather low activation energy barrier of 0.12 eV and is a thermodynamically spontaneous exothermic process. As a result, graphene-In2O3 exhibits superior electrocatalytic performance for the generation of urea. This work provides a revelation for subsequent researches that the mechanism of C—N coupling reactions may not be uniformly absolute, and may be flexible depending on the catalysts.
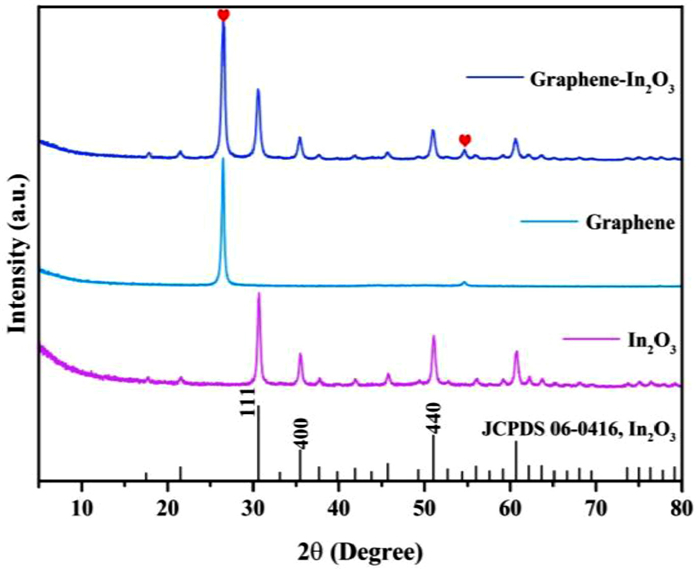
The cubic phase In2O3 with space group of la-3 (206) (a = b = c = 10.118 Å, Z = 16) were prepared by hydrothermal method and subsequent calcination [16]. As shown in Fig. 1, the X-ray diffraction pattern (XRD) of the prepared In2O3 is completely consistent with the standard card result (JCPDS No. 06–0416), and no other impurity peaks are observed. The cell parameters of In2O3 were calculated as a = b = c = 10.079 Å according to the measured XRD data, which is in agreement with the theoretical values and again demonstrates the high purity of the prepared In2O3 [17]. After the introduction of graphene (graphene-In2O3, graphene sheets were used as the catalyst supports), the XRD pattern of the sample shows only two additional C-peak as expected (marked by red hearts in Fig. 1), and the energy dispersive spectrum (EDS) in Fig. S2 (Supporting information) also demonstrates the successful synthesis of graphene-In2O3 (where Cu, Si, and some of C originate from the substrate). In particular, the characterization results of scanning electron microscopy (SEM) in Fig. 2a and Figs. S3b and c (Supporting information) reveal that the In2O3 nanoparticles are uniformly loaded on the graphene sheets.

|
Download:
|
| Fig. 1. XRD patterns of graphene-In2O3, graphene and In2O3. | |
{kind=link}

|
Download:
|
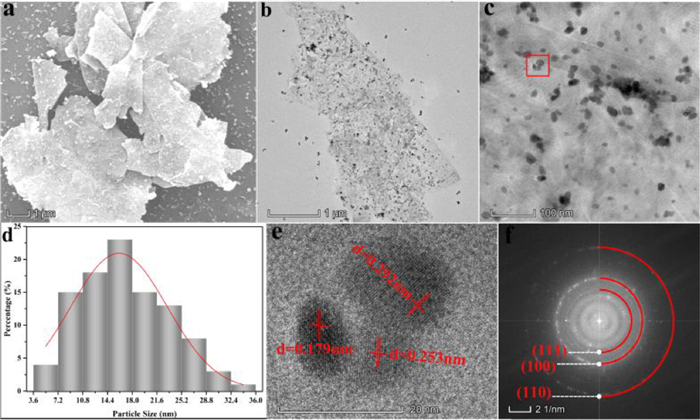
| Fig. 2. (a) SEM, (b) TEM, (c) high-magnification TEM, (d) nanoparticle distribution, (e) HRTEM and (f) SAED images of graphene-In2O3. | |
{kind=link}
Transmission electron microscopy (TEM) characterizations were used to further dissect the microstructure of graphene-In2O3, for which the images of TEM, high magnification TEM, nanoparticle distribution, high-resolution TEM (HRTEM) and selected area electron diffraction (SAED) are shown in Figs. 2b-f, respectively. Observationally, In2O3 nanoparticles with an average particle size of 17.8 nm (Figs. 2c and d) are indeed homogeneously loaded on graphene sheets (Figs. 2a and b). The HRTEM (Fig. 2e) collected on the sample surface (the area circled with a red box in Fig. 2c) shows lattice stripes in three directions with lattice spacing of d = 0.292, 0.253 and 0.179 nm (Fig. S4b in Supporting information), which correspond to the (111), (100) and (110) crystal planes of cubic phase In2O3, respectively. The exposed facets of red box area were also investigated by SAED (Fig. 2f), and the circular symmetrical SAED pattern consistently shows relatively clear diffraction rings of (111), (100) and (110) lattice planes of In2O3 (Fig. 2f). Meanwhile, the relevant elemental mapping analysis was carried out (Fig. S4a in Supporting information) and the distribution of C, In and O is relatively homogeneous, which also provides a support for the aforementioned conclusion that the In2O3 nanoparticles are uniformly loaded on graphene.
TG analysis was performed (50–600 ℃) to investigate the thermal stability of graphene-In2O3 (Fig. S5 in Supporting information). The minor mass loss below 100 ℃ is due to the release of adsorbed water and the mass loss from 100 ℃ to 200 ℃ is mainly due to the pyrolysis of the oxygen containing groups. Subsequently, the weight loss slows down and becomes constant as the temperature continued to rise, indicating that the samples are thermally stable. Fig. S6 (Supporting information) displays the nitrogen gas adsorption isotherms of graphene and graphene-In2O3 (Fig. S6a), together with the corresponding pore size distribution (Fig. S6b). The BET specific surface area of graphene-In2O3 is 27.60 m2/g, which is significantly larger than that of In2O3 (8.82 m2/g). Graphene-In2O3 displays exactly the same type of isotherm with graphene, indicating that their pore size distributions are close to each other. Correspondingly, both graphene and graphene-In2O3 show mesopores in the range of 2–50 nm.
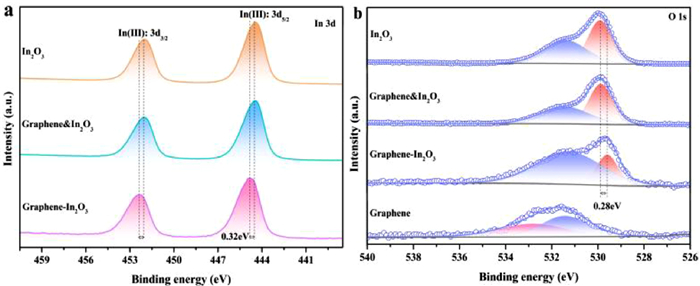
X-ray photoelectron spectroscopy (XPS) characterization was performed to further understand the surface chemical state of graphene-In2O3. The survey spectra of graphene & In2O3 (denotes a physical mixture of graphene and In2O3), graphene, graphene-In2O3 and In2O3 show no impurity peaks other than In, O, and C (Fig. S7 in Supporting information). Fig. 3a shows the high-resolution XPS spectra of In 3d core-level, two peaks at 444.4 and 452 eV are well-observed, which can be explained by the characteristic spin-orbit split 3d5/2 and 3d3/2, respectively, indicating the In3+ state [18, 19]. A slight upward shift in the binding energy of In to 444.72 and 452.32 eV can be observed in the graphene-In2O3 structure. In the meantime, the binding energies of O shift slightly downward from 531.48 eV to 529.9 eV (where the peak at high binding energy of 531.48 eV corresponds to the oxygen vacancies in the matrix of metal oxide and the peak at low binding energy of 529.9 eV is attributed to the oxygen bond of In-O-In [20]) to 531.2 and 529.62 eV (Fig. 3b). It has been shown that the number of oxygen vacancies here is very small and does not contribute significantly to the properties of the material (Figs. S8 and S9 in Supporting information). In addition, the peak at 533.2 eV of the graphene sample is attributed to hydroxyl oxygen on its surface [21]. High-resolution spectra of C 1s of graphene-In2O3, graphene and graphene & In2O3 were fitted to identify the carbon binding states (Fig. S10 in Supporting information), and four fitted peaks at 284.2, 284.8, 286.1 and 290.6 eV are obtained for all three samples, which are attributed to C—C, C = O, C—O and π-π* interactions, respectively [22].

|
Download:
|
| Fig. 3. High-resolution spectra of (a) In 3d and (b) O 1s. | |
{kind=link}
The Fourier transform infrared spectrum (FTIR) results are shown in Fig. S11 (Supporting information), several peaks in the range of 1650–1500 cm−1 together with 2800–2900 cm−1 are classified as the adsorbed water molecules; two peaks around 2300 cm−1 are attributed to the intercalated CO2 and the peak around 3400 cm−1 are caused by the O—H stretching vibrations [23]. While the presence of an absorption peak observed in the medium frequency area at 1045.42 cm−1 can be attributed to the stretching vibration of C—O bonds [21]. The peaks in the range of 600–400 cm−1 corresponding to the In-O phonon vibration are observed, which is the characteristic peak of the cubic In2O3 [23, 24]. Noticeably, both samples have three peaks at 540.06, 563.21 and 597.93 cm−1, where 540.06 cm−1 is caused by In-O stretching vibrations and 563.21 as well as 597.93 cm−1 are attributed to bending vibrations of In-O [16, 25]. Interestingly, the introduction of graphene resulted in two additional peaks (408.91 and 428.19 cm−1) in the infrared spectrum of the sample attributed to the bending vibrations of In-O [24], indicating that In2O3 was successfully immobilised on graphene, which explains the good dispersion of In2O3 on the graphene surface to a certain extent. Worthwhile, the In2O3 nanoparticles are anchored by In-O (surface O of graphene) rather than In-C (not detected), which together with the XPS results (O and In shifted respectively in the direction of low/high binding energies, while the binding energy of C showed no significant shift) suggests a weak electronic interaction between In2O3 and graphene. The Raman test was also performed (Fig. S12 in Supporting information), both samples show five peaks at 138, 314, 373, 503 and 635 cm−1, which are considered to be typical spectrum of cubic In2O3 [18, 26]. Of all the peaks, the peak at 138 and 373 cm−1 are individually attributed to the In-O vibration of InO6 structural units and the stretching vibrations of In-O-In, the peak at 314 and 503/635 cm−1 are usually interpreted as the bending vibration δ(InO6) and the stretching vibrations ν(InO6) of octahedrons, respectively [27-29]. Obviously, after the introduction of graphene, the In-O vibration is significantly enhanced, which is consistent with the results of FTIR.
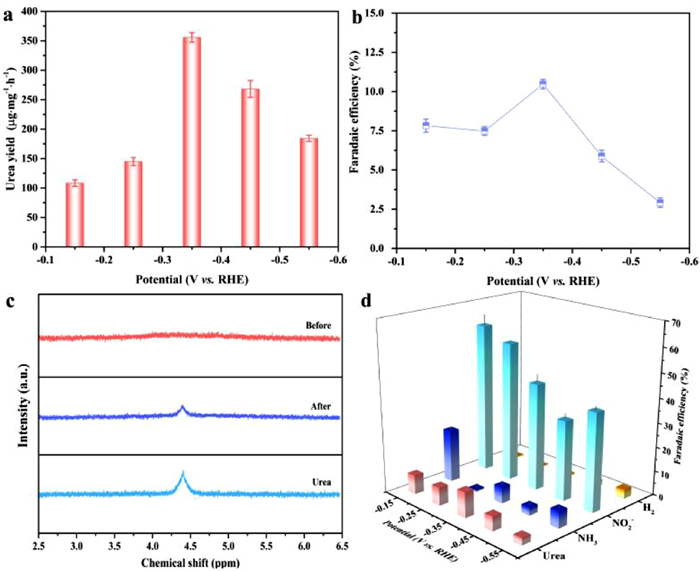
The performance of electrocatalytic urea synthesis of graphene-In2O3 was investigated in a gas-tight H-type electrolytic cell reactor using the Chrono-amperometry (CA) method at a range of fixed potentials (−0.15 ~ −0.55 V vs. RHE) in a mixed electrolyte of 5 mL 0.1 mol/L KNO3 and 15 mL 0.1 mol/L KHCO3 with a continuous feed of CO2 (see Methods in Supporting information). The quantification of each species was carried out by on-line gas chromatography (e.g., CO and H2) [19]. or the colorimetric methods (e.g., urea, NH3, and NO2−) (Figs. S13-S15 and Methods in Supporting information) [30]. The UV–vis spectrum as well as CA results (diacetylmonoxime method) for electrocatalytic urea synthesis (three times in all) are shown in Fig. S16 (Supporting information), where it can be noticed that a double peak phenomenon is evident compared to the standard peak (Fig. S13). According to the literature, this is most likely caused by the presence of NO2−, which not only distorts the peak but also brings about false positives. On the one hand, pure NO2− treated by the diacetylmonoxime method gives spectral results similar to those of urea; on the other hand, NO2− in coexistence with urea significantly increases the intensity of the spectrum of urea (Fig. S17 in Supporting information) [31]. Thus we used the urease method for a more accurate determination of urea concentration (Fig. S18 and Methods in Supporting information). Conspicuously, the average formation rate of urea and the FE reach the optimal values at −0.35 V vs. RHE, which are 357.47 µg mg−1 h−1 and 10.46%, respectively (Figs. 4a and b). It is noteworthy that the formation rate and FE only start to deteriorate slowly after six successive runs at the optimum potential (Figs. S19 and S20 in Supporting information). A long-term CA test (Fig. S21 in Supporting information) was also performed and the graphene-In2O3 is found to remain relative stability over a period of 40 h, as evidenced by the XRD and SEM results before and after the tests (Figs. S22 and S23 in Supporting information).

|
Download:
|
| Fig. 4. (a) Urea yields and (b) FE at different potentials (−0.15 ~ −0.55 V vs. RHE) in a mixed electrolyte of 5 mL 0.1 mol/L KNO3 and 15 mL 0.1 mol/L KHCO3 with a continuous feed of CO2. (c) 1H NMR spectra of electrolyte before and after electrolysis performance. (d) The FE for each product at different potentials (−0.15 ~ −0.55 V vs. RHE). | |
{kind=link}
The 1H NMR signals of the electrolyte before and after the electrocatalytic reaction suggests that urea is indeed produced by the electrocatalytic co-reduction of CO2 and NO3− (Fig. 4c). In addition to NH3, the NO2− by-product derived from NO3− were also identified (Figs. S24 and S25 in Supporting information), along with H2 production detected by gas chromatography (GC), and no other by-products (e.g., NH2OH, CO, N2) (Fig. S26 in Supporting information) were produced as probed by on-line gas chromatography and colorimetric methods. Based on the above results, the FE of each detected electrocatalytic product at different potentials is shown in Fig. 4d.
To further investigate the sources of elemental N and C in urea, a series of controlled experiments were carried out (Fig. S27 in Supporting information), and urea is found to be undetectable in the absence of either NO3− or HCO3−. Furthermore, the isotope-labeling electrocatalysis experiments were also performed in a mixed electrolyte of 5 mL 0.1 mol/L K15NO3 and 15 mL 0.1 mol/L KHCO3 with a continuous feed of CO2, and the peaks of 15N-labelled urea and NH3 were detected in the 1H NMR spectrum (Fig. S28 in Supporting information). Hence, the origin of C and N elements in urea are indeed derived from NO3− and HCO3−. Next, comparative experiments were launched based on the desire to understand the characteristics of the catalyst. Firstly, a verification experiment that carbon paper alone has no catalytic activity was performed (Fig. S29 in Supporting information), then the catalytic performance of graphene, In2O3, graphene-In2O3 and graphene & In2O3 for urea synthesis at −0.35 V vs. RHE in a mixed electrolyte of 5 mL 0.1 mol/L KNO3 and 15 mL 0.1 mol/L KHCO3 with a continuous feed of CO2 were investigated (Fig. S30 in Supporting information). Notably, the activity of graphene-In2O3 is much better than the other three for the electrocatalytic synthesis of urea, whereas In2O3 alone has observable activity and graphene alone is inactive. Considering the function of graphene as a promising carrier material that can improve the utilization of materials by increasing the specific surface area and dispersibility of the catalyst [32], together with the previous XPS discussion, it can be speculated that graphene enhances the activity of In2O3 mainly by homogeneous dispersion. However, the role of electron modulation cannot be completely ruled out. For this purpose, a comparative experiment was carried out to verify the speculated conclusions, and similar XPS results were obtained (Figs. S31-S36 in Supporting information) by replacing graphene with graphite (as evidenced by peak shifts in the high-resolution spectra of In 3d and O 1s), while graphite-In2O3 exhibits poorer catalytic performance due to the agglomeration of In2O3, which further illustrates that the graphene in graphene-In2O3 primarily exhibit the function of support and dispersant, thereby improving the adsorption behavior of the active sites in the electrocatalytic synthesis of urea. Additionally, the SEM results in Fig. S3 (Supporting information) also provide support for this conclusion. As shown in Fig. S3a, the separate In2O3 nanoparticles are heavily agglomerated, which greatly reduced the exposure of the active sites, resulting in low catalytic activity (crystals with smaller particle size tend to have larger surface energy, and in the absence of dispersing carrier, crystals with smaller particle sizes tend to aggregate, resulting in the occurrence of agglomeration). For graphene & In2O3, instead of being properly dispersed, the In2O3 are partially encapsulated by graphene (Figs. S3a and d), which further weakens the activity of the catalyst. Overall, graphene enhances catalytic activity by homogeneously dispersing In2O3 to increase the exposure of active sites, where the dispersion effect plays a key role.
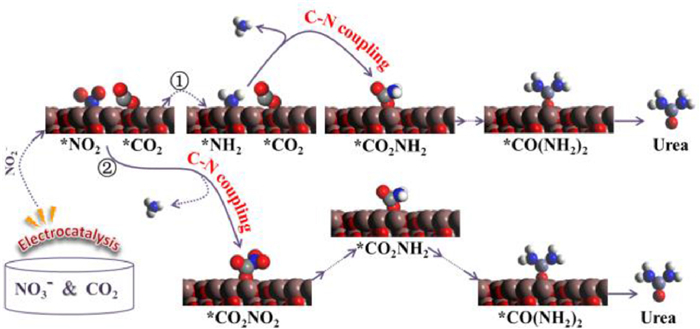
Subsequently, the mechanism of the C—N coupling reaction over graphene-In2O3 catalysts has been analysed. Various coupling mechanisms have been reported, such as *CO + *NH2, *COOH + *NH2, *CO2 + *NO2 and *CO + *N2 [12-17]. Worthwhile, NO2− and NH3 were detected in this work (Fig. 4d, Figs. S18 and 24), while no CO, N2 and NH2OH (Fig. S26) were detected during the entire reaction. In addition, the electrochemical reaction of NO3− to NH3 was represented by *NO3−→ *NO2 → *NO → *N → *NH → *NH2 → *NH3 or *NO3−→ *NO2 → *NO → *NOH → *HNOH → *NH2OH → *NH2 → *NH3 according to some previous reports [8, 33-35]. Since no production of NH2OH and CO were detected in the experiment, two possible reaction pathways are tentatively assumed from above analysis and available literatures [1, 8, 13, 36], either NO3− is first reduced to NO2− and further reduced to produce the key intermediate *NH2, which coupled directly with *CO2 to produce urea and the remainder combines with *H to produce NH3, or the *NO2 obtained from the reduction of NO3− coupled directly with *CO2 to produce urea (Fig. 5).

|
Download:
|
| Fig. 5. Two possible urea synthesis paths from CO2 and NO3− on graphene-In2O3 speculated by the experimental results and available literatures. | |
{kind=link}
Next, density functional theory (DFT) calculations were performed to further identify which of the inferred reaction mechanisms is responsible for the C—N coupling reaction on graphene-In2O3 electrocatalysis, and In2O3 (111) slab was modeled and calculated based on experimental and optimal adsorption configuration (Figs. 1 and 2e, Table S1 in Supporting information). The judgmental steps for urea production in the two alternative pathways are the coupling of *CO2 with *NH2 or *NO2, and the energies of their initial, final and transition states were separately calculated.
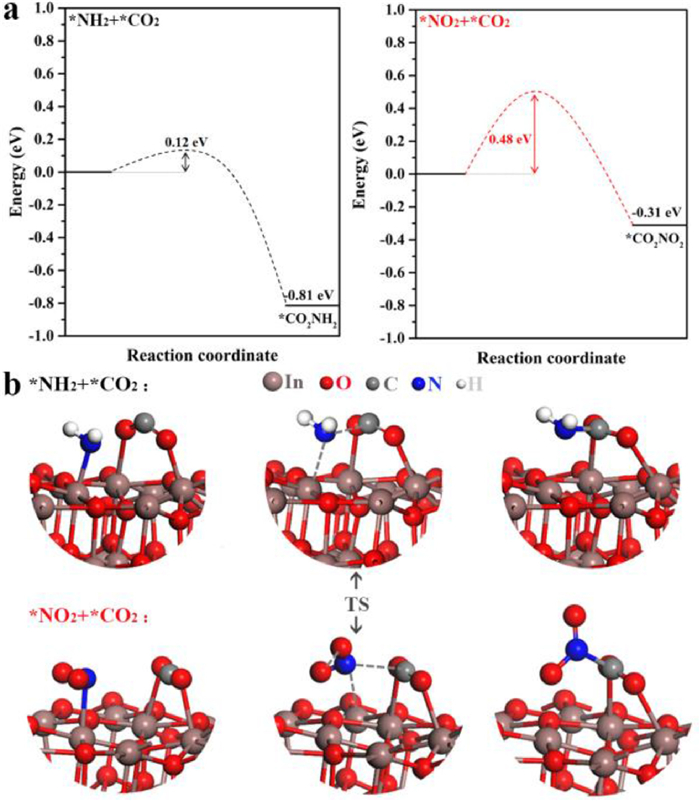
Fig. 6 shows separately the energy barriers in the C—N coupling pathways for the key intermediates and the corresponding transition state structures, the coupling reactions of *CO2 with *NH2 or *NO2 are both thermodynamically spontaneous processes with accompanying heat release values of −0.81 eV and −0.31 eV respectively. And the significant difference is reflected in the fact that the *CO2 + *NH2 →*CO2NH2 has a fairly low activation energy barrier of just 0.12 eV, while *CO2 + *NO2 → *CO2NO2 has an activation energy barrier of 0.48 eV, implying the possibility of direct coupling of *CO2 with *NH2 to produce urea over graphene-In2O3 catalysts is far more likely than that of *CO2 with *NO2.

|
Download:
|
| Fig. 6. (a) Energy barriers in the C—N coupling pathway of key intermediates. (b) The transition state structure for the *CO2 and *NH2 or *CO2 and *NO2 coupling. | |
{kind=link}
In order to demonstrate that *NH2 is the most probable key intermediate of the coupling reaction, a series of calculations were carried out. As shown in Figs. S37 and S38 (Supporting information), the energy barrier for direct coupling of *CO2 with *NOOH, *NO, *NOH, *N and *NH are respectively 0.38, 0.28, 0.24, 0.19 and 0.15 eV, which are all higher than that of *NH2 (0.12 eV), so CO2 should preferentially couple to NH2. Summarily, the C—N coupling reaction is more easily achieved in the *CO2 + *NH2 → *CO2NH2 process, i.e., here *CO2 and *NH2 act as key intermediates are coupled directly, a coupling mechanism that has not been reported previously. And the possible reaction pathway for co-reduction of carbon dioxide and nitrate to urea on graphene-In2O3 is proposed and illustrated in Fig. S39 (Supporting information). Thus, in combination with the experimental results of this work and what has been reported, we suggest that the key intermediates and reaction mechanisms for the C—N coupling reaction may vary flexibly with the catalysts.
In general, an eco-friendly and sustainable route for the high-efficiency production of urea has been developed in this work, and the designed graphene-loaded catalyst (graphene-In2O3) exhibits superior electrocatalytic performance for the generation of urea, with formation rate of 357.47 µg mg−1 h−1 and FE of 10.46% at −0.35 V vs. RHE. The functionality of graphene introduced during the catalyst synthesis has been analysed with the advantage of TEM and XPS based on a series of comparative experiments that were carried out, and the vigorous enhancement of the catalytic activity of In2O3 is primarily related to the dispersion effect of graphene, which fully exposes the active site of In2O3, facilitating the adsorption of CO2 and the reduction of NO3−, accelerating the electrocatalytic kinetics and promoting the formation of C—N bonds. Theoretical calculations and experiments synergistically demonstrate that the direct coupling of *CO2 and *NH2 in the C—N coupling reaction here possess an ultra-low energy barrier of 0.12 eV This work achieves a new way of thinking about the potential flexibility of key intermediates and reaction paths with changes in catalysts, which will be instructive for subsequent researches on electrocatalytic C—N coupling of urea and other important chemicals.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (No. 91741105), Chongqing Municipal Natural Science Foundation (No. cstc2018jcyjAX0625), and Program for Innovation Team Building at Institutions of Higher Education in Chongqing (No. CXTDX201601011).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108540.
| [1] |
Z. Tao, C.L. Rooney, Y. Liang, H. Wang, J. Am. Chem. Soc. 143 (2021) 19630-19642. DOI:10.1021/jacs.1c10714 |
| [2] |
D. Gao, H. Zhou, J. Wang, et al., J. Am. Chem. Soc. 137 (2015) 4288-4291. DOI:10.1021/jacs.5b00046 |
| [3] |
F. Naseem, P. Lu, J. Zeng, et al., ACS Nano 14 (2020) 7734-7759. DOI:10.1021/acsnano.0c02731 |
| [4] |
C. Chen, N. He, S. Wang, Small Sci. 1 (2021) 2100070. DOI:10.1002/smsc.202100070 |
| [5] |
D.B. Kayan, F. Köleli, Appl. Catal. B: Environ. 181 (2016) 88-93. DOI:10.1016/j.apcatb.2015.07.045 |
| [6] |
C. Chen, X. Zhu, X. Wen, et al., Nat. Chem. 12 (2020) 717-724. DOI:10.1038/s41557-020-0481-9 |
| [7] |
M. Yuan, J. Chen, Y. Bai, et al., Chem. Sci. 12 (2021) 6048-6058. DOI:10.1039/D1SC01467F |
| [8] |
Q. Hu, Y. Qin, X. Wang, et al., Energy Environ. Sci. 14 (2021) 4989-4997. DOI:10.1039/D1EE01731D |
| [9] |
M. Shibata, N. Furuya, Electrochim. Acta 48 (2003) 3953-3958. DOI:10.1016/S0013-4686(03)00534-6 |
| [10] |
M. Shibata, K. Yoshida, N. Furuya, J. Electroanal. Chem. 387 (1995) 143-145. DOI:10.1016/0022-0728(95)03992-P |
| [11] |
M. Shibata, K. Yoshida, N. Furuya, J. Electroanal. Chem. 442 (1998) 67-72. DOI:10.1016/S0022-0728(97)00504-4 |
| [12] |
M. Shibata, K. Yoshida, N. Furuya, J. Flectrochem. Soc. 145 (1998) 2348. DOI:10.1149/1.1838641 |
| [13] |
C. Lv, L. Zhong, H. Liu, et al., Nat. Sustain. 4 (2021) 868-876. DOI:10.1038/s41893-021-00741-3 |
| [14] |
N. Meng, Y. Huang, Y. Liu, Y. Yu, B. Zhang, Cell Rep. Phys. Sci. 2 (2021) 100378. DOI:10.1016/j.xcrp.2021.100378 |
| [15] |
J. Geng, S. Ji, M. Jin, et al., Angew. Chem. Int. Ed. 62 (2023) e202210958. DOI:10.1002/anie.202210958 |
| [16] |
F. Lei, Y. Sun, K. Liu, et al., J. Am. Chem. Soc. 136 (2014) 6826-6829. DOI:10.1021/ja501866r |
| [17] |
Y. Mao, Z. Wang, R. Ye, et al., Dalton Trans. 49 (2020) 15433-15442. DOI:10.1039/D0DT02955F |
| [18] |
A. Montazeri, F. Jamali-Sheini, Sens. Actuator. B 242 (2017) 778-791. DOI:10.1016/j.snb.2016.09.181 |
| [19] |
Z. Liang, L. Song, M. Sun, B. Huang, Y. Du, Nano Res. 16 (2023) 8757-8764. DOI:10.1007/s12274-023-5481-9 |
| [20] |
Y. Yang, Y. Ji, G. Li, et al., Angew. Chem. Int. Ed. 60 (2021) 26790-26797. DOI:10.1002/anie.202112042 |
| [21] |
M.K. R.abchinskii, S.D. Saveliev, D.Y. Stolyarova, et al., Carbon 182 (2021) 593-604. DOI:10.1016/j.carbon.2021.06.057 |
| [22] |
S. Park, Y. Hu, J.O. Hwang, et al., Nat. Commun. 3 (2012) 638. DOI:10.1038/ncomms1643 |
| [23] |
K.K. Pawar, L.S. Chaudhary, S.S. Mali, et al., J. Colloid Interface Sci. 561 (2020) 287-297. DOI:10.1016/j.jcis.2019.10.101 |
| [24] |
Z. Cai, M. Huang, J. Dai, et al., J. Colloid Interface Sci. 377 (2012) 347-354. DOI:10.1016/j.jcis.2012.03.072 |
| [25] |
B. Levasseur, A.M. Ebrahim, T.J. Bandosz, J. Colloid Interface Sci. 377 (2012) 347-354. DOI:10.1016/j.jcis.2012.03.072 |
| [26] |
L. Song, K. Dou, R. Wang, et al., ACS Appl. Mater. Interfaces 12 (2020) 1270-1279. DOI:10.1021/acsami.9b15928 |
| [27] |
L. Nagarajan, R.A. De Souza, D. Samuelis, et al., Nat. Mater. 7 (2008) 391-398. DOI:10.1038/nmat2164 |
| [28] |
N.M. Hung, N.M. Hieu, N.D. Chinh, et al., Sens. Actuator. B 313 (2020) 128001. DOI:10.1016/j.snb.2020.128001 |
| [29] |
A. Gurlo, D. Dzivenko, M. Andrade, et al., J. Am. Chem. Soc. 132 (2010) 12674-12678. DOI:10.1021/ja104278p |
| [30] |
M. Fu, Y. Mao, H. Wang, et al., Chin. Chem. Lett. 35 (2024) 108341. DOI:10.1016/j.cclet.2023.108341 |
| [31] |
X. Wei, X. Wen, Y. Liu, et al., J. Am. Chem. Soc. 144 (2022) 11530-11535. DOI:10.1021/jacs.2c03452 |
| [32] |
Y. Lu, L. Su, J. Lei, et al., J. Mater. Chem. A 6 (2018) 13717-13724. DOI:10.1039/C8TA03451F |
| [33] |
Y. Huang, R. Yang, C. Wang, et al., ACS Energy Lett. 7 (2021) 284-291. |
| [34] |
Y. Wang, A. Xu, Z. Wang, et al., J. Am. Chem. Soc. 142 (2020) 5702-5708. DOI:10.1021/jacs.9b13347 |
| [35] |
J.X. L.iu, D. Richards, N. Singh, B.R. Goldsmith, ACS Catal. 9 (2019) 7052-7064. DOI:10.1021/acscatal.9b02179 |
| [36] |
G. Wen, J. Liang, Q. Liu, et al., Nano Res. 15 (2021) 972-977. |