 2024, Vol. 35
2024, Vol. 35 


b State Key Laboratory of Microbial Technology, Shandong University, Qingdao 266237, China
More than 1000 labdane-type diterpenoids have been discovered as natural metabolites from various animals and plants [1–6], which enabled progresses in organic chemistry and disease treatment [7–9]. The deep-seated scaffold diversifications are derived from various pathways: biosynthetic, enzymatic, nonenzymatic, and postmodification pathways [10,11]. A series of intriguing labdane skeleton subtypes have emerged from the genus Pallavicinia of liverwort via oxidative cleavage, [4 + 2] cycloaddition, demethylation, and bond reconstruction of C2−C8, C4−C8, and C11−C3 as depicted in Supporting information (Fig. S1 in Supporting information) [12–14]. Organic chemists have shown an increasing interest in synthesizing these compounds in the past few decades [15–19].
During our ongoing research on these fascinating molecules from Pallavicinia ambigua (Mitt.) steph., we re-examined the components of P. ambigua collected from Hunan Province, China. Different molecules as depicted in Fig. 1, including two unprecedented cage-rigid 19-nor-7,8-seco-labdane diterpenoids pallaviambins A (1) and B (2), one derivative of the open tetrahydrofuran ring (7), and three dimeric congeners (8−10) formed via Diels–Alder (DA) cycloaddition, were identified via spectroscopic analyses, quantum chemical shift calculations, ECD calculations, and single-crystal X-ray diffraction measurements. The structures of the known labdane-type diterpenoids (3−6) [13] were also revised to be their corresponding enantiomers via ECD calculations, X-ray diffraction, and chemical conversion. Notably, architecturally intriguing labdanes 1 and 2 with tetracyclo[5.2.1.02,5.04,10]decane skeletons were formed via the [2 + 2] cycloaddition reaction of Δ1(2) and Δ6(7), verified by exposing congener 5 to ultraviolet (UV) light. The cytotoxic assay revealed that compound 5 significantly inhibited PC-3 cells with an IC50 value of 2.5 µmol/L via an apoptotic pathway. Herein, we describe the isolation, structural elucidation, photoinduced rearrangement, and cytotoxicity of these compounds.

|
Download:
|
| Fig. 1. Structures of compounds 1−10 isolated from P. ambigua. | |
{kind=link}
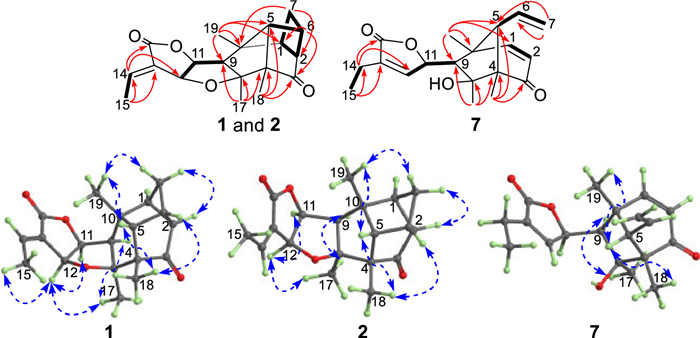
Pallaviambin A (1) was obtained as colorless needles, whose molecular formula was established as C19H22O4 based on the HRESIMS (m/z 337.1419, [M + Na]+), indicating nine double bond equivalents (DBEs). Additionally, its 1H NMR data (Table S1 in Supporting information) showed signals of four methyl groups [δH 1.07, s; 1.19, s; 1.22, s; and 2.06, dd (J = 7.2, 2.3 Hz)] and one olefinic proton [δH 7.10, qd (J = 7.2, 2.3 Hz)]. Furthermore, 19 carbons were assigned by combining the HMQC spectrum with the 13C NMR data (Table S2 in Supporting information), of which one ketone carbonyl (δC 215.7), one ester carbonyl (δC 170.6), and two oxygenated methines (δC 85.5 and 74.3) were identified. The NMR data of compound 1 were similar to that of 5 [13], except for the absence of two double bonds Δ1(2) and Δ6(7) and the upfield chemical shift values of C-1 (δC 55.5), C-2 (δC 61.7), C-6 (δC 41.9), and C-7 (δC 35.9). The same DBEs as compound 5 and the observed cross peaks for H-1/H2-7 and H-2/H-6 in the 1H−1H COSY spectrum suggested the formation of a new four-membered ring via unprecedented C1−C7 and C2−C6 linkages, as confirmed by the HMBC signals from H2-7 to C-2/C-10 and from H-6 to C-1/C-3 (Fig. 2). Therefore, compound 1 possessed a cage-rigid structure with a novel tetracyclo[5.2.1.02,5.04,10]decane skeleton, as depicted in Fig. 1.

|
Download:
|
| Fig. 2. Key HMBC (H→C), 1H−1H COSY (H▬H), and NOESY correlations (H↔H) of 1, 2, and 7. | |
{kind=link}
Furthermore, the signals of H-12/H-11 and H3-17, and H-9/H3-17 in the NOESY spectrum revealed that they are cofacial and designated to be α-oriented, whereas H-5/H3-18 and H3-19 are designated to be β-oriented. Finally, a crystal of compound 1 was obtained from MeOH. The subsequent single-crystal X-ray diffraction analysis, considering a Flack parameter of 0.07(6) and using CuKα radiation (CCDC 2083309), unequivocally determined the absolute structure as 1S, 2R, 4S, 5S, 6S, 8S, 9R, 10R, 11S and 12S.
Pallaviambin B (2) exhibited highly similar NMR data (Tables S1 and S2 in Supporting information) as compound 1, except that the resonances for H-14 and C-14 in it were shifted by −0.37 and 2.2 ppm in the 1H and 13C NMR data, respectively, owing to the anisotropic influence of the lactone carbonyl functional group [20,21]. Thus, compound 2 was speculated to be an E-Δ13(14) isomer of compound 1.
Moreover, compounds 1 and 2 were obtained simultaneously using the same methodology as that of photoinduced interconversion of compound 5 to compounds 3 and 4 [10]. Furthermore, [2 + 2] cycloaddition between Δ1(2) and Δ6(7) of 5 via free-radical reaction afforded compounds 1 and 2 (Scheme 1). Fresh P. ambigua was analyzed via high-performance liquid chromatography (Fig. S2 in Supporting information), which validated the natural occurrence of compounds 1 and 2.

|
Download:
|
| Scheme 1. The photo-driven conversion of compounds 1 and 2 from 5. | |
{kind=link}
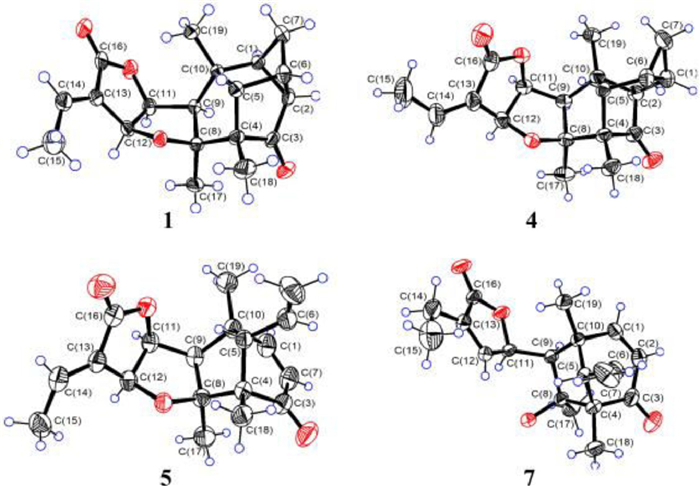
The original absolute configurations of known homologs 3−6 were established by CD exciton chirality method [13], which display limitations when the absorbed signal is weak or overlaps. Reinvestigation of the structure of compound 5 by determining its X-ray structure (CCDC 2191362) using CuKα radiation (Fig. 3), comparing calculated ECD curves with experimental ones (Fig. S3 in Supporting information), and combining with the already determined structures of 1 and 2, revised compound 5 and its isomer 6 as enantiomers of previously reported pallambins D and C, respectively [13]. Similarly, the X-ray structure of 4 (CCDC 2191361) (Fig. 3) and the ECD data (Fig. S3 in Supporting information) established the light-driven product 4 derived from compound 5 to be an enantiomer of pallambin A and compound 3 to be pallambin B accordingly [13].

|
Download:
|
| Fig. 3. X-ray crystallographic structures of compounds 1, 4, 5, and 7. | |
{kind=link}
Pallaviambin C (7), obtained in the form of colorless needles, yielded the molecular formula C19H24O4, as deduced via HRESIMS (m/z 339.1574, [M + Na]+), indicating one less DBE than compound 5. Comparing NMR data with that of compound 5 shows that the C8−O−C12 bond of the tetrahydrofuran ring was cleaved and the double bond (Δ12(13)) was endocyclic in compound 7. This was validated by the upfield chemical shift value of C-8 (δC 80.3) and downfield chemical shift value of C-12 (δC 147.3), as well as the key HMBC signals from H3-17 (δH 1.19, s) to C-8/C-9 (δC 58.6) and from H-14 [δH 2.33, q (J = 7.5 Hz)] to C-12/C-16 (δC 173.0) (Fig. 2). The absolute configuration (4S, 5S, 8S, 9R, 10R, and 11R) of compound 7 was conclusively determined by combining X-ray diffraction (CuKα radiation, CCDC 2205558) (Fig. 3) and ECD calculation (Fig. S4 in Supporting information).
Pallaviambin D (8), an amorphous powder, was assigned a molecular formula according to its HRESIMS peak at m/z 651.2933 [M + Na]+ (calculated for C38H44O8Na+, 651.2928). Comparing its 13C NMR data with that of compound 7 shows that compound 8 displayed 38 signals and appeared in pairs as follows: two keto-carbonyl groups (δC 201.8 and 201.2), four pairs of double bonds at ∆1(2) (δC 158.9, 127.0 and 158.8, 126.7) and ∆6(7) (δC 133. 1, 121.9 and 132.9, 121.6), three pairs of methyl groups (δC 29.0, 27.6; 19.8, 19.7; and 13.7, 13.6), and one pair of hydroxylated carbons (δC 81.4 and 80.7). These results indicated that compound 8 is a dimeric derivative of 7 and has the same bicyclo[3.2.1]octane frameworks (units A and B). The newly formed fragment of tetrahydro-1(3H)-isobenzofuranone (unit C) was determined based on the 1H–1H COSY cross-peaks of H-12/H-14′/H2-15′/H2-15/H-14 and HMBC correlations from H-11 [(δH 4.35, dd (J = 9.1, 2.0 Hz)] to C-12 (δC 42.4)/C-13 (δC 128.4)/C-14′ (δC 28.6); H-14 (δH 6.95, m) and C-16 (δC 169.1); and H-15′a (δH 2.01, m) and C-14 (δC 135.8) (Fig. S5A in Supporting information). The γ-lactone ring (unit D) linked to C-9′ was determined by the unassigned ether carbonyl (δC 174.1), ether carbon (δC 81.3), endocyclic double bond (δC 149.9 and 133.3), and HMBC correlations from H-12′ (δH 7.23, m) to C-9′ (δC 57.3)/C-16′. Finally, the 1H–1H COSY signal of H-9/H-11 and HMBC correlations from H-15′ to C-13′/C-14′ confirmed that unit C linked units A and D via C11–C9 and C14′–C13′, respectively. Accordingly, the planar structure of compound 8 was determined.
The relative configuration of compound 8 was determined from the NOESY spectrum. Units A and B exhibited the same NOESY signals as those of compound 7. For unit C, H-12 and H-14′ were axially oriented in the six-membered ring with a chair conformation, whereas H-11 was β-oriented, according to the NOESY correlations between H-9/H-12, H3-17, and H-14′ and H-11/H3-19. The small JH-9′, H-11′ (5.0 Hz) value and key NOESY correlation of H-11′/H3–19′, as demonstrated in the Newman projection formula (Fig. S5B in Supporting information), suggested the α-orientation of H-11′ in unit D in compound 8.
Pallaviambin E (9) had the same molecular formula as compound 8 base on HRESIMS. The 1D NMR data of 9 (Tables S3 and S4 in Supporting information) resembled those of 8 except that the resonances for H-9′, H-11′, and H-12′ in compound 9 were shifted by −0.61, −0.21 and 0.21 ppm, respectively; moreover, the resonances for C-9′, C-10′ and C-12′ were considerably low-field shifted by 0.9−5.9 ppm, compared with those for compound 8. The 2D NMR data revealed that compounds 8 and 9 had the same planar structure. Furthermore, the NOESY signals of units A, B, and C remained unchanged. However, unlike in the case of compound 8, the considerably large JH-9′, H-11′ (10.1 Hz) value and Newman projection formula along the C9′−C11′ bond (Fig. S5B in Supporting information) indicated the β-orientation of H-11′ in compound 9, as observed from the correlations between H-11′/H3-19′ and H-12′/H3-17′ in NOESY spectrum.
The above analysis of the NOESY correlations and coupling constants (JH-9′, H-11′) of compounds 8 and 9 strongly suggested the free rotation along C9′−C11′ bond was restricted due to the steric hindrance, and determined compounds 8 and 9 to be a pair of epimers at C-11′. To further validate their configurations, eight conformers of compound 8 (Fig. S6 in Supporting information) and six conformers of compound 9 (Fig. S7 in Supporting information) with a specific gravity of Boltzmann distribution > 1.0% were refined and considered for GIAO NMR shift calculation at the mPW1PW91/6–31G(d)//M062X/6–31G(d) level of theory. The correlation coefficient and CP3 [22] were the two parameters applied for quantifying the agreement between the calculated and experimental shifts. The results agreed with the NMR-based assignments for epimers 8 (11′R) and 9 (11′S) when the 1H and 13C NMR chemical shift values were considered (Figs. S8 and S9 in Supporting information). Finally, a good agreement of experimental and calculated ECD curves (Fig. S10 in Supporting information) determined the absolute configurations of compound 8 as 4S, 5S, 8S, 9R, 10R, 11R, 12R, 4′S, 5′S, 8′S, 9′R, 10′R, 11′R and 14′R; moreover, this agreement determined compound 9 as the 11′S epimer.
Pallaviambin F (10) was also found to be a labdane dimer with A and C units having similar spectroscopic data as compound 8. The NMR data of compound 10 indicated that its unit B is highly similar to that of 5, except for the replacement of the double bond Δ13′(14′) in compound 5 by two sp3 methine groups (δC 42.9 and 28.9) in compound 10, as interpreted via HMBC signals from H-13′ [δH 2.68, dd (J = 9.4, 3.5 Hz)] to C-12′ (δC 80.8)/C-14′/C-16′ (δC 176.5). Units B and C linked via the C13′−C14′ bond was identified via the 1H–1H COSY signals of H-13′/H-14′/H-12 and HMBC correlations from H-15′a (δH 1.79, m) to C-13′/C-14′ (Fig. S11 in Supporting information). Compound 10 shared the same relative stereochemistry concerning the unit A/C area with compound 8, as indicated by their similar 1D NMR and NOESY data. Considering the configuration of unit B, the NOESY signals of H-5′/H3-18′ and H3-19′ revealed that they were cofacial and β-oriented, whereas the observed correlations of H-12′/H-11′ and H3-17′, and H-9′/H3-17′ revealed that they were α-oriented. The newly generated chiral hydrogen (H-13′) was β-oriented, as determined from the NOESY signal of H-13′/H-11 and the absence of correlation with H-11′ and H-12′ (Fig. S11 in Supporting information). Furthermore, the experimental ECD spectrum (Fig. S10 in Supporting information) of compound 10 was in good accordance with the calculated one for the stereoisomer (4S, 5S, 8S, 9R, 10R, 11R, 12R, 4′S, 5′S, 8′S, 9′R, 10′R, 11′S, 12′S, 13′S and 14′R).
The formation of dimers 8‒10 can be explained by a biogenetic pathway. The key intermediate a, which might have formed from compound 7 via oxidation and keto–enol tautomerization, probably epimerizes to intermediates ⅰ (diene) and ⅱ (dienophile). Moreover, intermediate ⅱ cyclizes with intermediate ⅰ to form compounds 8 and 9 via the enzymatic DA cycloaddition. Additionally, compound 10 is likely to be formed via the DA cycloaddition of diene ⅰ and dienophile ⅲ. Furthermore, dienophile ⅲ might have been formed by the double bond isomerization of compound 5 (Scheme 2).

|
Download:
|
| Scheme 2. Plausible biogenetic pathway for compounds 1 and 8−10. | |
{kind=link}
Cytotoxic assay of isolated compounds with a panel of cell lines indicated that compounds 3−6 significantly inhibited the proliferation of PC-3 cells (Table S5 in Supporting information). We believe the electrophilic α,β-unsaturated ketone is crucial for cytotoxic activity, whereas the cleaved tetrahydrofuran ring might decrease the cytotoxicity of the examined compounds [5 (IC50 = 2.5 µmol/L) vs. 7 and 8 (IC50 > 20 µmol/L)]. The mechanism of action of compound 5 is shown in Fig. 4. A significant increase in the percentage of Annexin Ⅴ-positive cells was observed after dose-dependent treatment with compound 5. There was a marked increase in the expression of apoptosis-related proteins: Bax, cleaved caspase-3, and cleaved-PARP. This verified that compound 5 induces cell apoptosis in PC-3 cells. Approximately 60% of PC-3 cells were arrested in the G2/M phase after exposure to compound 5. The cellular reactive oxygen species (ROS) levels drastically increased in a dose-dependent manner, accompanied by a loss in mitochondrial membrane potential (MMP, ΔΨm) in the PC-3 cells. Thus, we believe that compound 5-treated cell death resulted from G2/M cell cycle arrest, triggering cellular apoptosis followed by damage to mitochondrial functions (increased ROS levels).

|
Download:
|
| Fig. 4. Compound 5 induces apoptosis in PC-3 cells. (A) Cells were collected and stained with Annexin Ⅴ and PI and analyzed via flow cytometry. (B) The expression of various apoptosis-related proteins was detected via Western blotting. PC-3 cells were treated with compound 5 for 24 h. (C) Determination of the cell cycle using different concentrations of compound 5 for 24 h. (D) The intracellular ROS level in PC-3 cells was detected by a DCFH-DA staining assay. Cells were exposed to compound 5 at several concentrations for 24 h. (E) Compound 5 induced the loss of ΔΨm in PC-3 cells after treatment at various concentrations and was measured via flow cytometry. These results are expressed as the mean ± SD (n = 3). **P < 0.1 and ***P < 0.001 vs. control. | |
{kind=link}
Collectively, compounds 1 and 2, a pair of cis–trans isomers with previously undescribed tetracyclo[5.2.1.02,5.04,10]decane scaffold, three DA adducts of labdane diterpenoids and their monomers (7−10) were isolated from P. ambigua. The formation of compounds 1 and 2 from compound 5 via light-driven [2 + 2] cycloaddition was demonstrated. Four previously reported homologous structures (3−6) were reinvestigated and revised as the corresponding enantiomers. Cytotoxicity assays indicated that compound 5 induced apoptosis through the accumulation of ROS in a PC-3 cell line and inhibited cell proliferation during the G2/M phase. Overall, our study shows that UV–vis light participates in the formation of complex structures, which enriches the chemical and bioactive diversity of labdane-type diterpenoids and simultaneously delivers several potential antitumor compounds.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this work.
AcknowledgmentsThis work is grateful to the staff of the research group for their dedication and national financial support from the National Key R&D Program of China (No. 2019YFA0905700), the National Natural Science Foundation of China (Nos. 82173703 and 81874293), and the Major Basic Research Program of Shandong Provincial Natural Science Foundation (No. ZR2019ZD26).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108206.
| [1] |
J.H. Langenheim, J. Chem. Ecol. 20 (1994) 1223-1280. DOI:10.1007/BF02059809 |
| [2] |
I. Rivero-Cruz, J.L. Trejo, M.I. Aguilar, R. Bye, R. Mata, Planta Med. 66 (2000) 734-739. DOI:10.1055/s-2000-9783 |
| [3] |
M. DellaGreca, A. Fiorentino, M. Isidori, P. Monaco, A. Zarrelli, Phytochemistry 55 (2000) 909-913. DOI:10.1016/S0031-9422(00)00253-3 |
| [4] |
M. Xu, M.L. Hillwig, S. Prisic, R.M. Coates, R.J. Peters, Plant J. 39 (2004) 309-318. DOI:10.1111/j.1365-313X.2004.02137.x |
| [5] |
I. Chinou, Curr. Med. Chem. 12 (2005) 1295-1317. DOI:10.2174/0929867054020990 |
| [6] |
A.K. Islam, O. Ohno, K. Suenaga, H. Kato-Noguchi, J. Plant Physiol. 171 (2014) 877-883. DOI:10.1016/j.jplph.2014.03.003 |
| [7] |
L.M.T. Frija, R.F.M. Frade, C.A.M. Afonso, Chem. Rev. 111 (2011) 4418-4452. DOI:10.1021/cr100258k |
| [8] |
J.R. Hanson, Nat. Prod. Rep. 32 (2015) 76-87. DOI:10.1039/C4NP00108G |
| [9] |
Q.T.N. Tran, W.S.F. Wong, C.L.L. Chai, Pharmacol. Res. 124 (2017) 43-63. DOI:10.1016/j.phrs.2017.07.019 |
| [10] |
J.Z. Zhang, R.X. Zhu, G. Li, et al., Org. Lett. 14 (2012) 5624-5627. DOI:10.1021/ol302295a |
| [11] |
J.C. Zhou, J.Z. Zhang, A.X. Cheng, et al., Org. Lett. 17 (2015) 3560-3563. DOI:10.1021/acs.orglett.5b01664 |
| [12] |
M. Toyota, T. Saito, Y. Asakawa, Chem. Pharm. Bull. 46 (1998) 178-180. DOI:10.1248/cpb.46.178 |
| [13] |
L.N. Wang, J.Z. Zhang, X. Li, et al., Org. Lett. 14 (2012) 1102-1105. DOI:10.1021/ol3000124 |
| [14] |
Y. Li, Z.J. Xu, R.X. Zhu, et al., Org. Lett. 22 (2020) 510-514. DOI:10.1021/acs.orglett.9b04270 |
| [15] |
J.Q. Dong, H.N.C. Wong, Angew. Chem. Int. Ed. 48 (2009) 2351-2354. DOI:10.1002/anie.200806335 |
| [16] |
C. Ebner, E.M. Carreira, Angew. Chem. Int. Ed. 54 (2015) 11227-11230. DOI:10.1002/anie.201505126 |
| [17] |
B. Huang, L. Guo, Y. Jia, Angew. Chem. Int. Ed. 54 (2015) 13599-13603. DOI:10.1002/anie.201506575 |
| [18] |
L.P. Martinez, S. Umemiya, S.E. Wengryniuk, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 7536-7539. DOI:10.1021/jacs.6b04816 |
| [19] |
X. Zhang, X. Cai, B. Huang, et al., Angew. Chem. Int. Ed. 58 (2019) 13380-13384. DOI:10.1002/anie.201907523 |
| [20] |
M. Nair, R. Adams, J. Am. Chem. Soc. 83 (1961) 922-926. DOI:10.1021/ja01465a041 |
| [21] |
T.S. Kam, Y.M. Choo, Phytochemistry 65 (2004) 603-608. DOI:10.1016/j.phytochem.2003.12.014 |
| [22] |
C.S. Kim, L. Subedi, J. Oh, et al., J. Nat. Prod. 80 (2017) 1134-1140. DOI:10.1021/acs.jnatprod.7b00111 |