 2024, Vol. 35
2024, Vol. 35 


b School of Pharmacy, Henan University, Kaifeng 475004, China;
c Department of Public Health, Chengdu Medical College, Chengdu 610500, China
Photothermal therapy (PTT) generates heat through photothermal conversion to increase the local temperature of the tumor, thus triggering thermal ablation of tumor cells [1,2]. Compared with photodynamic therapy or chemodynamic therapy, the efficacy of PTT is not limited by endogenous factors such as the content of oxygen or hydrogen peroxide in the tumor [3–7], and PTT possesses accurate spatiotemporal selectivity, which can minimize the damage to normal tissues [8–10]. While treating local tumors, PTT can not only rapidly kill tumor cells, resulting in cell apoptosis and necrosis, but also induce immunogenic cell death (ICD) and produce large amounts of tumor-associated antigens (TAAs), thus improving tumor immunogenicity [8,11,12]. Therefore, PTT can not only directly destroy the primary tumor, but also activate the anti-tumor immune response to inhibit tumor metastasis [13,14]. However, due to the heat endurance of tumor cells, PTT alone cannot completely eliminate tumor cells [15], and its inefficient ICD induction cannot produce strong anti-tumor immunity [14,16]. Therefore, combination with other therapy modes may effectively enhance the anti-tumor effect of PTT [17,18].
Hollow copper sulfide nanoparticles (HCuS) are popular materials in PTT due to their advantages of high photothermal conversion efficiency, low preparation cost, simple process, and degradation after laser irradiation, and HCuS are easy to load drugs, so they are widely used in combination therapy [19,20]. Disulfiram (DSF) is a commonly used anti-alcoholism drug in clinic, and recent studies have proven that DSF has Cu2+ dependent anti-tumor effect [21–23]. DSF is metabolized into diethyldithiocarbamate (DDTC) in vivo, which could be complexed with Cu2+ to form Cu(DDTC)2, thus inducing apoptosis and ICD of tumor cells and enhancing the immunogenicity of tumor cells [24–27]. However, the limited supply of Cu2+ in the body limits the application of DDTC [28]. Owing to the properties of near infrared (NIR) responded degradation, HCuS could release Cu2+ under laser irradiation [11], thus combining DDTC with HCuS may be an effective way to solve the copper source issue. In this way, HCuS could not only kill tumor cells through photothermal effect, but also release Cu2+ to form cytotoxic Cu(DDTC)2, which further acts on the residual heat tolerance cells, induces tumor cell apoptosis and ICD, and synergically enhances the immunogenicity of tumor cells.
However, how to increase the accumulation of photothermal agents at the tumor site is the key to enhancing the anti-tumor effect of PTT. Although nanoparticles can target solid tumor sites through the enhanced permeability and retention (EPR) effect of tumors, this passive targeting is inefficient [29]. In the design of nanosystems, ligands with high affinity for specific over-expressed receptors on tumor vascular endothelial cells or tumor cells are often modified on the surface of nanosystems, giving nanoparticles the ability to actively target tumors, thus enhancing tumor accumulation [30–34]. Hyaluronic acid (HA) is a commonly used cell-targeting material that could specifically bind to over-expressed CD44 receptors on the membrane of tumor cells, increasing the accumulation of HA-modified nanomaterials at tumor sites [35–38]. As an endogenous macromolecule, HA showed good biocompatibility. Thus modifying HA on the surface of nanoparticles could reduce the probability of nanoparticles being phagocytosed by the reticuloendothelial system (RES), and improve the blood circulation capacity of nanosystems [39,40].
The penetration of photothermal agents into tumor sites is an important but neglected step in the design of nanodrug delivery systems for PTT. PTT plays the role of tumor thermal ablation through the photothermal conversion of photothermal agents, thus the concentration and distribution of photothermal agents in tumor during irradiation directly affect the anti-tumor efficacy of PTT. However, due to the dense extracellular matrix (ECM) of tumors [41–43], the nanoparticles arriving at the tumor site are often distributed in the margin area and difficult to penetrate deeply, which leads to uneven distribution of photothermal and poor therapeutic effect. Tumor ECM mainly consists of various collagen, glycosaminoglycan, and growth factor molecules, which could support tumor cell growth, therapeutic resistance and metastasis [44,45]. Among these components, collagen accounts for about 90% of the ECM, and its expression level is positively correlated with interstitial resistance [46], which is the main barrier limiting tumor penetration. Losartan is a commonly used angiotensin Ⅱ receptor antagonist in clinical. In recent years, some studies have found that losartan could mediate the downregulation of tumor transforming growth factor β1 (TGF-β1) through angiotensin Ⅱ receptor type Ⅰ [47,48], reduce tumor collagen expression and improve the penetration of nanoparticles in tumors [45,49–52], so as to solve the problem of uneven photothermal distribution. Losartan can not only promote the penetration of nanoparticles but also increase the infiltration of immune cells in solid tumors [53–55], so as to enhance the immune killing effect of cytotoxic T lymphocytes (CTLs) on the primary and metastatic tumors. Therefore, losartan can be used as a tumor microenvironment regulator to enhance the efficacy of PTT against tumors and metastasis.
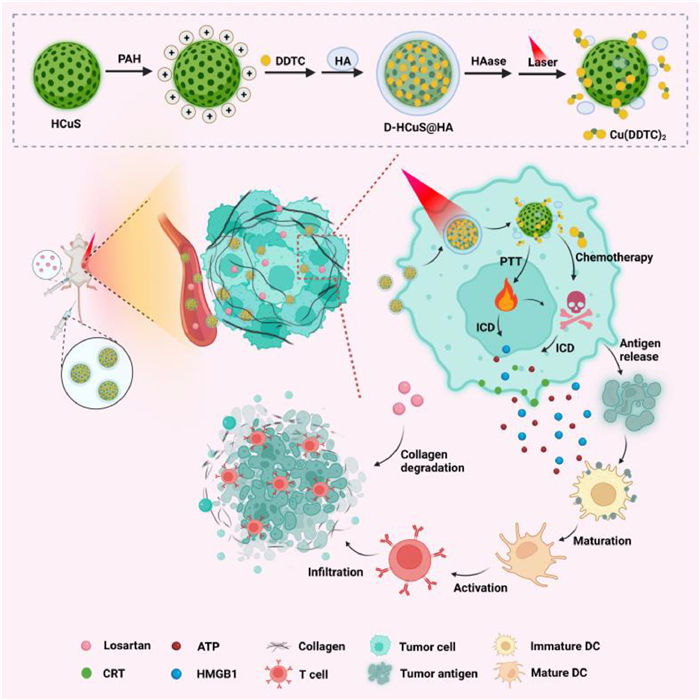
Along this line, we constructed a HA modified, DDTC loaded hollow copper sulfide nanoparticle D-HCuS@HA and combined it with losartan for the synergistic treatment of breast cancer and metastasis. As illustrated in Scheme 1, D-HCuS@HA could target tumor cells with the modification of HA to improve the tumor accumulation of nanoparticles, and further combined with losartan to improve the penetration of D-HCuS@HA at the tumor site by reducing tumor matrix density. Then the D-HCuS@HA, which evenly distributes in tumor tissue, could increase the local temperature of the tumor in response to laser irradiation, and release Cu2+, complexing with DDTC to form toxic Cu(DDTC)2, thus eliminating primary breast cancer through PTT and chemotherapy. Subsequently, TAAs and damage associated molecular patterns (DAMPs) produced in the process of primary tumor treatment promote the maturation of dendritic cells (DCs), activate the anti-tumor immune response, and inhibit the metastasis of breast cancer. Moreover, because losartan reduced tumor matrix density and improved the tumor microenvironment, CTL could infiltrate into the primary and metastatic tumors to play the immune effect, and comprehensively inhibit the development of breast cancer and metastases.

|
Download:
|
| Scheme 1. Schematic illustration of the construction of D-HCuS@HA, along with the anti-tumor mechanism of losartan combined with D-HCuS@HA. | |
{kind=link}
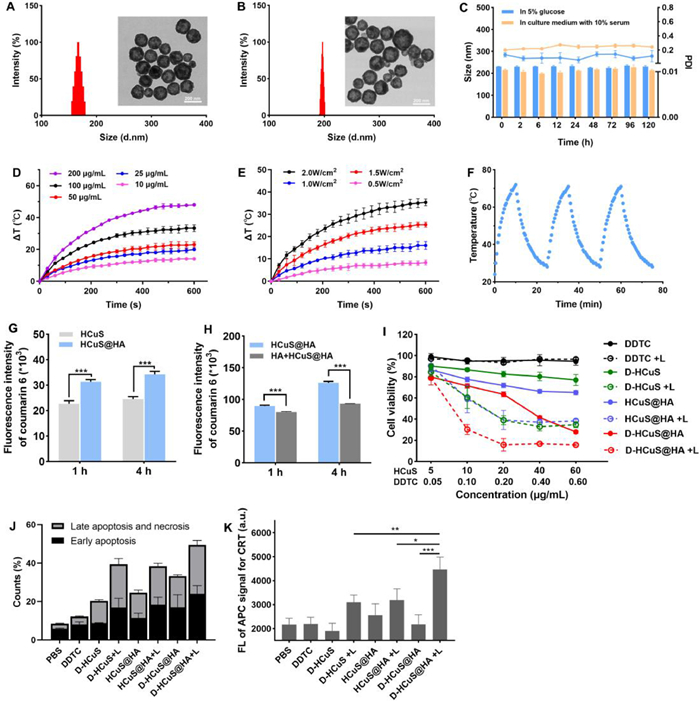
The construction of D-HCuS@HA is depicted in Scheme 1. Firstly, HCuS was prepared by a simple soft-template method as reported previously [11]. As shown in Fig. S1 (Supporting information), the typical peaks of Cu 2p at 931.6 and 951.5 eV and obvious S 2p peak at 161.5 eV in the X-ray photoelectron spectroscopy (XPS) spectrum of HCuS indicated that the main component of the synthesized HCuS was copper sulfide [56]. The transmission electron microscope (TEM) and dynamic light scattering (DLS) showed that the HCuS were spherical and uniform, with the hydrodynamic diameter of 177.9 ± 3.1 nm (Fig. 1A). The surface of HCuS was negatively charged (−13.4 ± 1.7 mV), and after cationization by PAH, the zeta potential of HCuS@PAH changed to 14.0 ± 2.4 mV (Fig. S2 in Supporting information). Then with the electrostatic adsorption of HA, the hydrodynamic diameter of HCuS@HA increased from 180.2 ± 3.2 nm (HCuS@PAH) to 191.4 ± 4.6 nm, and the surface charge converted to −20.9 ± 2.3 mV, indicating the successful modification of HA (Fig. S2). Next, to investigate the drug loading capacity of HCuS, we prepared D-HCuS@HA with different mass ratios of HCuS and DDTC, and the supernatants were collected to measure unloaded DDTC. In the UV–vis absorption curve (Fig. S3A in Supporting information), the characteristic absorption peak at 282 nm could be ascribed to DDTC, and free HA did not affect the absorption peak of DDTC. The supernatants of D-HCuS@HA with different mass ratios had no absorption peak at 282 nm and their absorption curves were coincidence with the supernatant of HCuS@HA (100:0), indicating that DDTC was totally loaded in D-HCuS@HA. DLS measurements showed that with the increasing amount of DDTC, the PDI of D-HCuS@HA increased slightly, and the diameter increased to 324.1 ± 10.6 nm (Fig. S3B in Supporting information), indicating that the higher ratio of DDTC might affect the dispersibility of D-HCuS@HA. Then we determined the stability of D-HCuS@HA with different DDTC loading ratios in aqueous solution. As shown in Fig. S3C (Supporting information), when D-HCuS@HA at the mass ratio of higher than 40:1, the particle size increased rapidly, manifesting that the higher ratio of DDTC could reduce the stability of nanoparticles and lead to the aggregation of D-HCuS@HA. Therefore, the mass ratio of 100:1 was chosen for the subsequent experiments. As shown in Fig. 1B, the obtained D-HCuS@HA exhibited uniform morphology with a hydrodynamic diameter of 202.3 ± 3.3 nm. Next, the stability of D-HCuS@HA in different media was monitored at certain times by DLS. The particle size and PDI of D-HCuS@HA showed ignorable change after incubating at 5% glucose or culture medium with 10% serum for 120 h, suggesting good colloidal stability (Fig. 1C).

|
Download:
|
| Fig. 1. Characterizations of nanoparticles and the and synergistic therapeutic effects in vitro. Size distribution measured by DLS and TEM image of (A) HCuS and (B) D-HCuS@HA. (C) Particle size and PDI changes of D-HCuS@HA in different media for 120 h (n = 3). Photothermal heating curves of D-HCuS@HA solutions at (D) different concentrations and (E) different laser efficiencies (n = 3). (F) Photothermal stability of D-HCuS@HA (n = 3). (G) Quantitative uptake of Cou6 loaded HCuS and Cou6 loaded HCuS@HA by 4T1 cells. (H) Quantitative uptake of Cou6 loaded HCuS@HA by 4T1 cells before and after HA preincubation. (I) MTT cytotoxicity of different preparations in laser free and laser (+L) state. (J) The quantitative analysis of apoptosis. (K) Quantitative analysis of CRT expression on 4T1 cells treated with different preparations by flow cytometry (n = 3). Data are presented as means ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001. | |
{kind=link}
The photothermal properties of D-HCuS@HA were tested using the 808 nm laser. As shown in Fig. 1D, the temperature of D-HCuS@HA solutions increased with the extension of irradiation time, and the terminal temperature reached higher with the increase of D-HCuS@HA concentrations. The enhanced power density of irradiation could also make D-HCuS@HA solution reach a higher temperature (Fig. 1E). The temperature of 100 µg/mL D-HCuS@HA increased 35.3 ℃ under 2 W/cm2 irradiation in 10 min, and reached a terminal temperature of 60.3 ℃. The photothermal stability was further evaluated, and the temperature curve of cyclic photothermal conversions of D-HCuS@HA showed that the peak temperatures in three cycles did not decrease, suggesting that the photothermal conversion ability of D-HCuS@HA was stable (Fig. 1F). All the data manifested that D-HCuS@HA could induce a rapid and stable photothermal effect.
Then HCuS were labeled with coumarin 6 (Cou6) to evaluate the intracellular uptake efficacy of different nanoparticles using flow cytometry assay and fluorescence microscope imaging. As shown in Fig. 1G, the uptake of HCuS and HCuS@HA by 4T1 cells increased over time, indicating that the uptake of nanoparticles was time-dependent. It was worth noting that the fluorescence signals of HCuS@HA group were 1.39-fold higher than that of HCuS group after incubation for 4 h. Then we used HA pre-incubated 4T1 cells to further investigated the effect of HA-modifying on HCuS uptake. As shown in Fig. 1H, the fluorescence signal of HCuS@HA was 1.35-fold lower in HA pre-incubated 4T1 cells than that of normal 4T1 cells, that is because pre-incubation with HA could saturate the CD44 receptor on 4T1, thus reducing the specific binding between CD44 and HCuS@HA. The results of fluorescence microscope imaging (Fig. S4 in Supporting information) were consistent with the flow cytometry result, suggesting that HA modification could enhance the uptake of HCuS in 4T1 cells, which is conducive to the ablation of tumors.
The synergistic effect of chemo-PTT of D-HCuS@HA was evaluated on tumor cytotoxicity. According to the MTT assay, the viabilities of 4T1 cells remained at high rates after treating with free DDTC or DDTC+L (Fig. 1I), and the cell viability of HCuS@HA group was over 70% even at a high dose, indicating that DDTC and HCuS had no obvious toxicity on cells. However, the viability of cells in HCuS@HA+L group obviously decreased compared to that of HCuS@HA, suggesting HCuS could induce the ablation of cells under irradiation. Notably, D-HCuS@HA+L group showed the lowest cell viability, which could due to the synergistic effect of PTT and the complexation of DDTC and Cu2+. Under laser irradiation, the Cu2+ from degraded HCuS could be chelated with DDTC, resulting in the active complex of Cu(DDTC)2. Therefore, the cytotoxicity of low-dose D-HCuS@HA was significantly enhanced. In addition, losartan showed no toxicity to 4T1 cells and did not affect the cytotoxicity of D-HCuS@HA under irradiation (Fig. 1I and Fig. S5 in Supporting information). Then according to the Annexin Ⅴ-fluorescein isothiocyanate/propidium iodide (Annexin Ⅴ-FITC/PI) staining, the proportion of both early apoptotic cells and late apoptotic cells in D-HCuS+L, HCuS@HA+L, and D-HCuS@HA+L groups were significantly increased compared with their non-laser counterparts (Fig. 1J and Fig. S6 in Supporting information), indicating that PTT could rapidly induce apoptosis of 4T1 cells. It is worth noting that D-HCuS@HA+L resulted in the severest cell apoptosis, whose apoptosis percentage was 1.49-fold higher than that of HCuS@HA+L, suggesting that D-HCuS@HA could further enhance the apoptosis through Cu(DDTC)2 toxicity. Similar results were obtained in calcein-AM/PI staining (Fig. S7 in Supporting information). D-HCuS@HA+L group exhibited the strongest red fluorescence, indicating the greatest cell-killing effect. These results jointly manifested that DDTC loaded in D-HCuS@HA endows HCuS with better anti-tumor efficacy, which proved the effectiveness of the PTT-chemotherapy.
As the typical markers of ICD, calreticulin (CRT) translocation, high mobility group protein B1 (HMGB-1), and ATP release were detected to investigate the synergistic ICD effect. The CRT translocation was detected using flow cytometry assay and fluorescence microscope imaging. As shown in Fig. S8 (Supporting information), D-HCuS+L, HCuS@HA+L, and D-HCuS@HA+L groups exhibited more CRT translocation than non-laser groups, suggesting PTT could induce obvious CRT translocation. Quantitative results of CRT exposure on 4T1 cells were shown in Fig. 1K, which were consistent with the above results. Notably, the fluorescence intensity of CRT in D-HCuS@HA+L group is 1.44-fold and 1.41-fold higher than that of D-HCuS+L group and HCuS@HA+L group, respectively. Although free DDTC could not increase the CRT translocation, the D-HCuS@HA+L induced more CRT translocation than that of HCuS@HA+L, indicating that Cu(DDTC)2 could amply CRT translocation. Moreover, D-HCuS@HA+L also induced the most ATP and HMGB-1 release (Figs. S9 and S10 in Supporting information). These data jointly manifested that DDTC loading and HA modification could collaborate with PTT and further improve the ICD effect, which would mature more DCs and activate stronger anti-tumor immune response.
Next, HCuS and HCuS@HA were labeled with Cou6 to detect the distribution of nanoparticles in tumor tissues, all animal experiments were performed under the guidance and approved by the Ethics Committee of Sichuan University (Chengdu, China). Based on the excellent NIR absorption capacity of HCuS, photoacoustic (PA) imaging was used for the in vivo tracking of nanoparticles in the tumor site. As shown in Fig. S11 (Supporting information), the PA signals of tumors in the two groups administrated with HCuS@HA were significantly stronger than that in the HCuS group at 2 h post-injection, suggesting that modifying HA on the surface of HCuS could increase the accumulation of nanoparticles at the tumor site. The PA signals tended to vanish at 24 h post-injection, indicating that the peak time of nanoparticles at the tumor site was less than 24 h after administration. Ex vivo tumor sections in Fig. 2A showed that the fluorescence intensity of collagen Ⅰ in the tumor margin area was significantly decreased in Losartan+HCuS@HA group, demonstrating that intraperitoneal injection of losartan could reduce the collagen distribution in the tumor site, thereby reducing the density of tumor matrix. The fluorescence distribution of Cou6 in tumor margins in the HCuS@HA and Losartan+HCuS@HA groups was significantly enhanced compared with that in the HCuS group, suggesting that HA modification can increase the targeting ability of HCuS to 4T1 tumors. It is worth noting that in the tumor sections of Losartan+HCuS@HA group, the Cou6 fluorescence was not only confined to the tumor margin but also showed more distribution of fluorescence signals at a distance from the tumor margin, indicating that in the mice treated with losartan, the HCuS@HA could cross the tumor matrix and penetrate the tumor depth. The biodistribution of nanoparticles in ex vivo organs was detected by Cu concentration using atomic absorption spectrophotometry. As shown in Fig. S12 (Supporting information), the liver tissue showed the highest Cu concentration, while less distribution of Cu was detected in the kidney, indicating that HCuS and HCuS@HA nanoparticles were metabolized and cleared mainly through the liver. In addition, the Cu concentrations in tumors of HCuS@HA and Losartan+HCuS@HA groups were about 1.20-fold higher than that in HCuS group, suggesting that HA modification could increase the targeting effect of HCuS on tumors.

|
Download:
|
| Fig. 2. Targeted accumulation and penetration and the anti-tumor effect in vivo. (A) Distribution of Cou6-loaded nanoparticles and collagen Ⅰ at the ex vivo tumor margin area and center area. Scale bar: 100 µm. (B) Tumor growth curves and (C) survival times of 4T1-bearing mice (n = 6). (D) Bioluminescence imaging of postoperative 4T1-luc metastasis mice at certain times. (E) Ex vivo lung tissues and its H&E staining sections of postoperative 4T1-luc metastasis mice. Red arrows represent pulmonary metastatic nodules. Scale bar: 200 µm. Data are presented as means ± SD. | |
{kind=link}
To further determine the time of peak concentration of D-HCuS@HA at tumor sites after administration, we detected the changes in Cu concentration in tumor tissues at different time points post-injection. As shown in Fig. S13 (Supporting information), the tumor Cu concentration of mice injected with D-HCuS@HA was higher than that of mice injected with D-HCuS during the whole determination time, which further demonstrated that HA modification could increase the targeting effect of HCuS on 4T1 tumors, which was consistent with the results of PA imaging. The Cu concentration of the tumor in D-HCuS group decreased gradually over time, while in the two groups treated with D-HCuS@HA, the Cu concentration of the tumor increased first and reached the peak at 6 h post-injection, then decreased slowly. In addition, at 6 h post-injection, the Cu concentration of the tumor in Losartan+D-HCuS@HA group was higher than that of mice injected with D-HCuS@HA alone, further supporting that losartan can increase the penetration of D-HCuS@HA at the tumor site, thus increasing the accumulation of nanoparticles. Here we determined the peak time of nanoparticles, thus subsequent laser irradiation of PTT would be carried out on tumor sites at 6 h after administration.
Next, thermal imaging was performed to investigate the targeting and photothermal conversion capacity of D-HCuS@HA in vivo. As shown in Fig. S14A (Supporting information), the temperature of tumor sites increased gradually under irradiation. After 5 min of laser irradiation, the local tumor temperature of mice in Losartan+D-HCuS@HA group reached 59.7 ℃, showing the best photothermal effect. According to the photothermal temperature variation curve in Fig. S14B (Supporting information), the temperature increases faster in D-HCuS@HA and Losartan + D-HCuS@HA group compared with that of D-HCuS group. These data once again manifested that HA modification on the surface of HCuS could improve the targeting capacity of HCuS, and losartan could further increase the accumulation of D-HCuS@HA at the tumor site, thus improving the photothermal effect and precisely ablating tumor cells.
The potential anti-tumor and anti-metastasis efficacy of losartan combined with D-HCuS@HA upon 808 nm laser irradiation was further investigated on female BALB/c mice bearing 4T1 tumors. As shown in Fig. 2B, the tumor volume of mice in control group (administrated with 5% glucose) and DDTC group increased rapidly with the extension of time, and Losartan+D-HCuS@HA showed ignorable tumor suppression. All mice in the above three groups were considered dead due to tumor volume exceeding 1500 mm3 before day 26 (Fig. 2C), suggesting that administration of D-HCuS@HA without laser irradiation did not produce anti-tumor effects, nor did losartan. Mice treated with PTT showed significant tumor regression, and 91.67% of the tumors in the four laser irradiation groups were measured to be completely ablated at day 14. However, some tumors in PTT groups began to recur on day 18. As shown in Fig. 2B, the tumor volume of mice in D-HCuS+L group and HCuS@HA+L groups showed rapid growth, while in D-HCuS@HA+L and Losartan+D-HCuS@HA+L groups the tumor volume grew slowly, indicating that the toxicity of Cu(DDTC)2 and the modification of HA could significantly enhance the anti-tumor effect of HCuS. The body weights of mice in all groups were increased during the treatment, indicating the good biosafety of these preparations (Fig. S15 in Supporting information).
The metastasis model was established by intravenous injection of 4T1-luc cells (5 × 105) into mice after surgical removal of the orthotopic tumor. Bioluminescence imaging was used to monitor the development of metastasis in vivo on certain days. As shown in Fig. 2D, severe lung metastases were observed in 5% glucose, DDTC, and Losartan+D-HCuS@HA groups. The bioluminescence signals were enhanced over time, indicating a gradual increase in metastases in the mice, leading to their death. Some mice in D-HCuS+L group and HCuS@HA +L group also showed transfer signals at day 18, while no obvious transfer signals were found in D-HCuS@HA+L group and Losartan+D-HCuS@HA+L group compared with the previous five groups. A large number of pulmonary nodules appeared in 5% glucose, DDTC, and Losartan+D-HCuS@HA groups (Fig. S16 in Supporting information), which was consistent with the bioluminescence signals. The pulmonary nodule number in four PTT groups decreased significantly, among which the Losartan+D-HCuS@HA+L group showed the least nodules with the inhibition rate of lung metastasis reaching 95.38%. The hematoxylin and eosin (H&E) staining of lung tissues also showed similar results (Fig. 2E). These data indicate that losartan combined with D-HCuS@HA under irradiation could not only inhibit primary tumor but also suppress the development of postoperative metastases, which manifested that the anti-tumor immunity is activated in the treated mice.
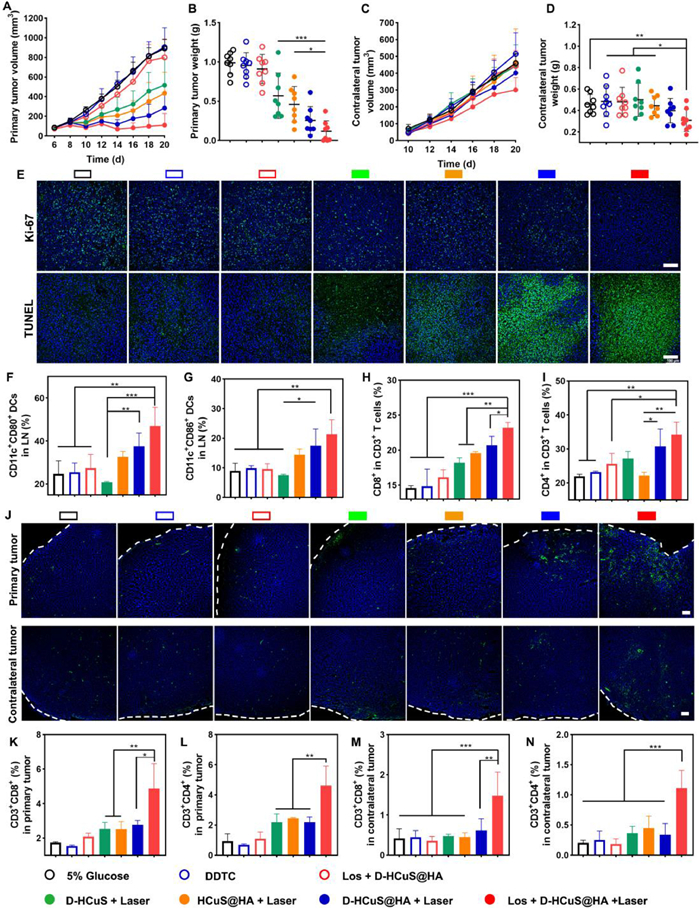
Given the finding that D-HCuS@HA+L could induce ICD of tumor cells, and Losartan+D-HCuS@HA+L could suppress tumor metastases in vivo, we speculated that losartan combined with D-HCuS@HA upon 808 nm laser irradiation could effectively activate immune response. To further determine the mechanism of the synergistic anti-tumor effect of the combined therapy, we next studied the anti-tumor efficacy of losartan combined with D-HCuS@HA upon 808 nm laser irradiation on the bilateral 4T1 orthotopic tumor models. The volume of primary tumor in control (administrated with 5% glucose), DDTC and Losartan+D-HCuS@HA groups increased rapidly (Fig. 3A), which was consistent with the results in orthotopic 4T1 tumor model. As the laser irradiation time in the therapeutic schedule of this model was shortened from 5 min to 2 min, in which the intensity of PTT was reduced, the volume of the primary tumor at D-HCuS+L and HCuS@HA+L groups gradually increased with the extension of time. The mean weights of the primary tumor in D-HCuS+L and HCuS@HA+L groups were 2.19-fold and 1.76-fold of that in D-HCuS@HA+L group (Fig. 3B), respectively, which manifested that the combination strategy of chemo-PTT and HA-modified targeting strategy could improve the anti-tumor efficacy of HCuS. The lowest tumor volume and weights of both primary (Figs. 3A and B) and contralateral tumors (Figs. 3C and D) were detected in Losartan+D-HCuS@HA+L group, displaying the best anti-tumor efficacy. Then H&E, Masson, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL), and Ki-67 staining of primary and contralateral tumors were carried out (Fig. 3E, Figs. S17 and S18 in Supporting information). There were more nuclei damage and degradation in the Losartan+D-HCuS@HA+L group compared with other groups in primary and contralateral tumor slices, along with the strongest expression of apoptosis signal and the weakest Ki-67 signal, indicating that losartan combined with D-HCuS@HA upon 808 nm laser irradiation could effectively kill tumor cells, induce cell apoptosis and reduce tumor proliferation. Masson staining showed that the expression of collagen fibers (stained blue in the slices) in the tumor matrix was significantly reduced in groups treated with losartan. Losartan had been demonstrated to improve the penetration of nanoparticles, which ensures photothermal evenly distribute in the primary tumor during laser irradiating, thus enhancing the anti-tumor effect. These results jointly illustrated the advantages of losartan enhanced chemo-PTT and HA-driven targeting, which suppressed the development of primary tumors and distant metastatic tumors.

|
Download:
|
| Fig. 3. Anti-tumor effect and the immune response in bilateral tumor mice in vivo. (A) Tumor growth curves and (B) tumor weights of the primary tumors (n = 8). (C) Tumor growth curves and (D) tumor weights of the contralateral tumors (n = 8). (E) Ki-67 immunofluorescence and TUNEL staining of the primary tumor sections. The proportion of (F) CD11c+CD80+ mature DCs and (G) CD11c+CD86+ mature DCs in LNs (n = 3). The proportion of activated (H) CD8+ T cells and (I) CD4+ T cells in CD3+ T cells in LNs (n = 3). (J) Immunofluorescent staining of effector T cell (staining marker was CD8α) in bilateral tumor sections, the white dotted line represents the tumor edge. The proportion of (K) CD3+CD8+ T cells (L) CD3+CD4+ T cells in the primary tumor (n = 3). The proportion of (M) CD3+CD8+ T cells (N) CD3+CD4+ T cells in the contralateral tumor (n = 3). Scale bars represent 100 µm. Data are presented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. | |
{kind=link}
Next, the biosafety of these preparations was investigated. No significant changes in body weights were observed (Fig. S19 in Supporting information), illustrating little acute toxicity of these preparations. The H&E staining of major organs illustrated that there was no significant pathological damage in all groups (Fig. S20 in Supporting information). In addition, the serological and hematological assessment were further carried out. The serum enzyme detection of all groups showed that the liver function, renal function, and heart function indexes including alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), creatinine (CREA), urea (UREA), uric acid (UA), creatine kinase (CK) were controlled within the normal ranges (Fig. S21 in Supporting information). The routine blood tests showed that the relevant indexes of red blood cells and platelets were normal in all groups (Fig. S22 in Supporting information), but the white blood cells count (WBC) decreased significantly in groups treated by PTT, which was closer to the normal level, especially in Losartan+D-HCuS@HA+L group, indicating that the inflammation of tumor-bearing mice was significantly improved after Losartan+D-HCuS@HA+L treatment. All of these results demonstrated that the therapeutic regimen of losartan combined with D-HCuS@HA upon laser irradiation could reduce the inflammation level in tumor-bearing mice without causing obvious damage, and had good biological safety.
Then, we assessed the immune response of bilateral orthotopic tumor-bearing mice. First, the primary tumor slices were stained with immunofluorescence to detect exposure and release of the specific DAMPs. As shown in Fig. S23 (Supporting information), Losartan+D-HCuS@HA+L remarkably promoted CRT and HMGB-1 expression in primary tumor tissue, which improved the immunogenicity of primary tumors. Then mature DCs in lymph nodes and spleen were detected by flow cytometry. Consistent with the capacity to induce ICD, there was the highest percentage of CD80+CD86+ DCs in Losartan+D-HCuS@HA+L treated mice (Fig. S24 in Supporting information). The proportion of CD80+ DCs and CD86+ DCs in the lymph nodes of Losartan+D-HCuS@HA+L treated mice were 1.91-fold and 2.38-fold of the control group (Figs. 3F and G), and similar results were also observed in the spleen (Fig. S25 in Supporting information), suggesting that losartan combined with D-HCuS@HA upon irradiation could effectively promote the maturation of DCs. Mature DCs could further stimulate and activate T cells in lymph nodes to induce anti-tumor immune responses, so we then tested the levels of activated T cells in lymph nodes. The proportions of CD3+CD8+ T cells and CD3+CD4+ T cells in Losartan+D-HCuS@HA+L group were the highest in all groups (Figs. S26 and S27 in Supporting information), and the proportions of activated CD4+ or CD8+ cells in CD3+ T cells were also significantly increased (Figs. 3H and I), suggesting that losartan combined with D-HCuS@HA upon irradiation could stimulate T cell activation and increase the ratio of helper T cells and effector T cells, thereby amplify the immune response. Then we detected the infiltration of activated T cells in bilateral tumors. As shown in the immunofluorescence staining of bilateral tumors (Fig. 3J), CD8α signals were significantly enhanced in the primary and contralateral tumor sections in Losartan+D-HCuS@HA+L group, indicating an increase of effector T cells in the tumor. To quantitatively detect T cell infiltration, flow cytometry was used to measure the proportions of CD3+CD8+ T cells and CD3+CD4+ T cells in bilateral tumors. In primary tumors, the proportions of CD3+CD8+ T cells and CD3+CD4+ T cells increased in all irradiation groups (Figs. 3K and L), suggesting that PTT could induce anti-tumor immune response in primary tumors. Notably, Losartan+D-HCuS@HA+L group showed a significant increase in the proportion of activated T cells in primary tumors. Compared with D-HCuS@HA+L group, the proportion of CD3+CD8+ T cells increased from 2.78% to 4.87%, CD3+CD4+ T cells increased from 2.18% to 4.61% in the primary tumor of Losartan+D-HCuS@HA+L group. The increasing proportions of activated T cells were also observed in the contralateral tumor of Losartan+D-HCuS@HA+L group (Figs. 3M and N). The proportion of CD3+CD8+ T cells and CD3+CD4+ T cells increased to 3.56-fold and 5.49-fold in Losartan+D-HCuS@HA+L group compared with the control group. However, no obvious increase of infiltrating T cells was observed in the contralateral tumor of D-HCuS@HA+L group. As a distant metastatic tumor that did not receive direct PTT, the therapeutic efficacy of the contralateral tumor mainly depended on the anti-tumor immune response induced by in situ therapy. In the Losartan+D-HCuS@HA+L group, losartan could decrease the expression of tumor collagen, and improve T cell infiltrating, thus causing anti-tumor effect on the distant metastatic tumor and inhibiting their growth. Although the D-HCuS@HA+L group greatly inhibited the growth of the primary tumor and activated anti-tumor immune response in the host, the activated T cells could not infiltrate into the distant metastatic tumor due to the dense tumor matrix, thus producing ignorable contralateral tumor suppression.
Briefly, these results jointly verified that the preparation with HA-driven targeting capacity and losartan enhanced chemo-PTT could trigger potent ICD of tumor cells and remodel the immunosuppressive microenvironment, which promotes DC maturation, T cell activation, and infiltration to exert effective anti-tumor and anti-metastasis efficacy.
In this work, we designed a HA-driven and light-controlled drug delivery platform D-HCuS@HA combined with losartan for synergistic anti-tumor therapy. Through HA-driven active targeting, D-HCuS@HA could target the tumor sites in which CD44 receptors were over-expressed. In vitro and in vivo experiments demonstrated that losartan could decrease the density of the tumor matrix to improve the accumulation and penetration of nanoparticles in the tumor site. Then D-HCuS@HA could achieve a nontoxic to toxic transformation within the tumor, in which the laser irradiation would lead to photothermal conversion to exert the PTT effect, and meanwhile release Cu2+ to chelate the loaded DDTC, generating cytotoxic Cu(DDTC)2 for effective killing of tumor cells while avoiding systemic toxicity. Significantly, PTT and chemotherapy both induced cell apoptosis and ICD of tumor cells to trigger potent systemic anti-tumor immune response. Moreover, the therapeutic efficacy of D-HCuS@HA upon irradiation could be significantly enhanced when combined with losartan to modulate the immunosuppressive microenvironment, which improved the infiltration of activated T cells in primary and distant metastatic tumors. Therefore, our findings provided a new strategy for enhancing PTT therapy by utilizing the properties of nanomaterials to synergize tumor cell killing and anti-tumor immune response, and in combination with losartan to improve the immunosuppressive microenvironment and increase T cell infiltration, to realize the synergistic photothermal-chemo-immune therapy and inhibit the development and metastasis of breast cancer.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (No. 82173762), Research Funds of Sichuan Science and Technology Department (Nos. 2022JDJQ0050, 2022YFS0334) and 111 Project (No. B18035).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108765.
| [1] |
H. Wang, L. Gao, T. Fan, et al., ACS Appl. Mater. Interfaces 13 (2021) 54621-54647. DOI:10.1021/acsami.1c13634 |
| [2] |
Q.W. Chen, X.H. Liu, J.X. Fan, et al., Adv. Funct. Mater. 30 (2020) 1909806. DOI:10.1002/adfm.201909806 |
| [3] |
X. Zhang, S. Wang, G. Cheng, et al., Engineering 13 (2022) 18-30. DOI:10.1016/j.eng.2021.07.023 |
| [4] |
Y. Zhou, F. Tong, W. Gu, et al., Acta Pharm. Sin. B 12 (2022) 1416-1431. DOI:10.1016/j.apsb.2021.12.001 |
| [5] |
S. Li, F. Yang, Y. Wang, et al., Chem. Eng. J. 451 (2023) 138621. DOI:10.1016/j.cej.2022.138621 |
| [6] |
J.X. Fan, M.Y. Peng, H. Wang, et al., Adv. Mater. 31 (2019) 1808278. DOI:10.1002/adma.201808278 |
| [7] |
Z. Tang, P. Zhao, H. Wang, et al., Chem. Rev. 121 (2021) 1981-2019. DOI:10.1021/acs.chemrev.0c00977 |
| [8] |
J. Li, Y. Zhou, J. Liu, et al., J. Control. Release 352 (2022) 313-327. DOI:10.1016/j.jconrel.2022.10.025 |
| [9] |
H.S. Jung, P. Verwilst, A. Sharma, et al., Chem. Soc. Rev. 47 (2018) 2280-2297. DOI:10.1039/C7CS00522A |
| [10] |
R. Liu, C. Luo, Z. Pang, et al., Chin. Chem. Lett. 34 (2023) 107518. DOI:10.1016/j.cclet.2022.05.032 |
| [11] |
F. Tong, H. Hu, Y. Xu, et al., Acta Pharm. Sin. B 13 (2023) 3471-3488. DOI:10.1016/j.apsb.2022.11.003 |
| [12] |
J. Qin, T. Yang, J. Li, et al., Nano Today 46 (2022) 101591. DOI:10.1016/j.nantod.2022.101591 |
| [13] |
T. Shang, X. Yu, S. Han, B. Yang, Biomater. Sci. 8 (2020) 5241-5259. DOI:10.1039/D0BM01158D |
| [14] |
Y. Chen, P. He, D. Jana, et al., Adv. Mater. 34 (2022) 2201706. DOI:10.1002/adma.202201706 |
| [15] |
H. Chen, B. Ding, J. Tan, et al., Chem. Eng. J. 442 (2022) 136296. DOI:10.1016/j.cej.2022.136296 |
| [16] |
Z. Chen, Q. Zhang, Q. Huang, et al., Int. J. Pharm. 617 (2022) 121578. DOI:10.1016/j.ijpharm.2022.121578 |
| [17] |
R. Liu, W. Xiao, C. Hu, et al., J. Control. Release 278 (2018) 127-139. DOI:10.1016/j.jconrel.2018.04.005 |
| [18] |
C. Hu, X. Cun, S. Ruan, et al., Biomaterials 168 (2018) 64-75. DOI:10.1016/j.biomaterials.2018.03.046 |
| [19] |
A.N. Nikam, A. Pandey, G. Fernandes, et al., Coord. Chem. Rev. 419 (2020) 213356. DOI:10.1016/j.ccr.2020.213356 |
| [20] |
D. Wang, H. Dong, M. Li, et al., ACS Nano 12 (2018) 5241-5252. DOI:10.1021/acsnano.7b08355 |
| [21] |
Z. Skrott, M. Mistrik, K.K. Andersen, et al., Nature 552 (2017) 194-199. DOI:10.1038/nature25016 |
| [22] |
D. Chen, Q.C. Cui, H. Yang, Q.P. Dou, Cancer Res. 66 (2006) 10425-10433. DOI:10.1158/0008-5472.CAN-06-2126 |
| [23] |
W. Wu, L. Yu, Y. Pu, et al., Adv. Mater. 32 (2020) 2000542. DOI:10.1002/adma.202000542 |
| [24] |
X. Li, K. Du, J. Sun, F. Feng, ACS Appl. Bio Mater. 3 (2020) 654-663. DOI:10.1021/acsabm.9b01014 |
| [25] |
W. Chen, W. Yang, P. Chen, et al., ACS Appl. Mater. Interfaces 10 (2018) 41118-41128. DOI:10.1021/acsami.8b14940 |
| [26] |
X. Gao, H. Huang, C. Pan, et al., Cancers 14 (2022) 4715. DOI:10.3390/cancers14194715 |
| [27] |
Q. Li, Y. Chao, B. Liu, et al., Biomaterials 291 (2022) 121880. DOI:10.1016/j.biomaterials.2022.121880 |
| [28] |
H. Tang, C. Liu, J. Zhang, et al., ACS Appl. Mater. Interfaces 12 (2020) 47289-47298. DOI:10.1021/acsami.0c14636 |
| [29] |
R. Liu, C. Hu, Y. Yang, et al., Acta Pharm. Sin. B 9 (2019) 410-420. DOI:10.1016/j.apsb.2018.09.001 |
| [30] |
X. Yang, C. Hu, F. Tong, et al., Adv. Funct. Mater. 29 (2019) 1901896. DOI:10.1002/adfm.201901896 |
| [31] |
Y. Tie, H. Zheng, Z. He, et al., Signal Transduct. Target. Ther. 5 (2020) 6. DOI:10.1038/s41392-020-0115-0 |
| [32] |
S. Ruan, W. Xiao, C. Hu, et al., ACS Appl. Mater. Interfaces 9 (2017) 20348-20360. DOI:10.1021/acsami.7b02303 |
| [33] |
N.U. Khan, J. Ni, X. Ju, et al., Acta Pharm. Sin. B 11 (2021) 1341-1354. DOI:10.1016/j.apsb.2020.10.015 |
| [34] |
J. Ni, T. Miao, M. Su, et al., J. Control. Release 329 (2021) 934-947. DOI:10.1016/j.jconrel.2020.10.023 |
| [35] |
Z. Luo, Y. Dai, H. Gao, Acta Pharm. Sin. B 9 (2019) 1099-1112. DOI:10.1016/j.apsb.2019.06.004 |
| [36] |
O. Gotov, G. Battogtokh, Y.T. Ko, Mol. Pharm. 15 (2018) 4668-4676. DOI:10.1021/acs.molpharmaceut.8b00640 |
| [37] |
M. Zhang, C. Xu, L. Wen, et al., Cancer Res. 76 (2016) 7208-7218. DOI:10.1158/0008-5472.CAN-16-1681 |
| [38] |
J. Sun, J. Li, X. Li, et al., Chin. Chem. Lett. 34 (2023) 107891. DOI:10.1016/j.cclet.2022.107891 |
| [39] |
M. Wen, N. Yu, S. Wu, et al., Bioact. Mater. 18 (2022) 242-253. |
| [40] |
S. Sharma, J. Singh, A. Verma, et al., RSC Adv. 6 (2016) 73083-73095. DOI:10.1039/C6RA11260A |
| [41] |
W. Yu, C. Hu, H. Gao, Adv. Drug Deliv. Rev. 178 (2021) 113909. DOI:10.1016/j.addr.2021.113909 |
| [42] |
X. Gu, Y. Gao, P. Wang, et al., J. Control. Release 333 (2021) 374-390. DOI:10.1016/j.jconrel.2021.03.039 |
| [43] |
J. Insua-Rodríguez, T. Oskarsson, Adv. Drug Deliv. Rev. 97 (2016) 41-55. DOI:10.1016/j.addr.2015.12.017 |
| [44] |
H. Yang, Z. Tong, S. Sun, Z. Mao, J. Control. Release 328 (2020) 28-44. DOI:10.1016/j.jconrel.2020.08.024 |
| [45] |
S. Yang, H. Gao, Pharmacol. Res. 126 (2017) 97-108. DOI:10.1016/j.phrs.2017.05.004 |
| [46] |
P.A. Netti, D.A. Berk, M.A. Swartz, et al., Cancer Res. 60 (2000) 2497-2503. |
| [47] |
J.E. Murphy, J.Y.L. Wo, D.P. Ryan, et al., J. Clin. Oncol. 36 (2018) 4116. DOI:10.1200/JCO.2018.36.15_suppl.4116 |
| [48] |
A. García-Martín, C. Navarrete, M. Garrido-Rodríguez, et al., Biomed. Pharmacother. 142 (2021) 112007. DOI:10.1016/j.biopha.2021.112007 |
| [49] |
B. Diop-Frimpong, V.P. Chauhan, S. Krane, et al., Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 2909-2914. DOI:10.1073/pnas.1018892108 |
| [50] |
Y. Tang, Y. Liu, S. Wang, et al., Drug Deliv. Transl. Res. 9 (2019) 615-624. DOI:10.1007/s13346-018-00610-1 |
| [51] |
J. Hu, X. Yuan, F. Wang, et al., Chin. Chem. Lett. 32 (2021) 1341-1347. DOI:10.1016/j.cclet.2020.11.006 |
| [52] |
X. Cun, S. Ruan, J. Chen, et al., Acta Biomater. 31 (2016) 186-196. DOI:10.1016/j.actbio.2015.12.002 |
| [53] |
S. Liu, M. Zhang, H. Yu, et al., ACS Appl. Mater. Interfaces 15 (2023) 7700-7712. DOI:10.1021/acsami.2c18371 |
| [54] |
Y. Sun, W. Zhao, D.R. Spriggs, et al., Clin. Cancer Res. 26 (2020) PR07. |
| [55] |
Y. Xu, J. Xiong, X. Sun, H. Gao, Acta Pharm. Sin. B 12 (2022) 4327-4347. DOI:10.1016/j.apsb.2022.11.001 |
| [56] |
J. Peng, X. Lu, X. Jiang, et al., Chem. Eng. J. 354 (2018) 740-752. DOI:10.1016/j.cej.2018.08.038 |