 2024, Vol. 35
2024, Vol. 35 


b Department of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou 350122, China;
c Shanghai Wei Er Lab, Shanghai 201707, China;
d Shanghai Skin Disease Hospital, Tongji University School of Medicine, Shanghai 200443, China
Breast carcinoma is one of the most devastating malignant tumors in women with high morbidity and mortality [1,2]. Pursuing effective therapies for breast carcinoma is highly important for improving lifespan and living quality of patients. Other than surgery, chemotherapy remains the mainstream treatment for breast carcinoma, especially unresectable and metastasis ones. Among numerous first-line chemotherapeutic agents, paclitaxel (PTX) is mostly utilized owing to its high and broad-spectrum antitumor efficacy. By acting on the microtubule system, PTX can block mitosis and arrest cells in the G2/M phase [3,4].
PTX has a dose-dependent antitumor pattern, and its efficacy is always hampered by its poor solubility. To address this issue, various drug carriers have been developed, such as micelles, nanoparticles, emulsions, liposomes to solubilize PTX and elevate the dose levels [5]. The most representative formulation is based on polyoxyethylene castor oil (Cremophor EL) micelles, and has been approved by the Food and Drug Administration (FDA) for over 30 years with a brand name of Taxol®. However, high irritation and toxicity caused by Cremophor EL always requires a corticosteroid premedication, limiting the maximum tolerated dose (MTD) and compromising the efficacy of PTX. Consequently, its domination in the global market was weakened after the approval of Genexol-PM® (PTX polymeric micelles) and Abraxane® (albumin-bound PTX nanoparticles) which have lower toxicity, higher MTD, and superior efficacy [6,7]. Nowadays, lots of other PTX carriers are emerging, aiming at further potentiating efficacy by enhanced MTD and drug delivery efficiency, as well as decreasing expenses [8,9].
Among various drug carriers, liposomes are one kind of the most successful vehicles which have paved the way to market, owing to its excellent biocompatibility, biodegradability, drug loading, and delivery efficiency [10,11]. And their versatile surface characteristics introduced by various synthetic lipids enable multi-functionalities. For example, a PEGylated liposomal formulation (Doxil®) confers doxorubicin long circulation, reduced cardiac toxicity, and enhanced tumor accumulation [12,13]. PTX can also be loaded into liposomes by incorporation into the lipid-bilayers. Liposomal PTX has been developed and approved in China for the first-line chemotherapy for patients with ovarian carcinoma, non-small cell lung cancer and breast carcinoma with a brand name of Lipusu® [14-16]. Nevertheless, Lipusu® suffers from low stability, low drug loading, and unsatisfied efficacy, owing to rapid drug leakage and precipitation of PTX [17-20]. To improve the compatibility between PTX and liposomes, derivatization of PTX by lipids has been widely adopted, by which high and stable encapsulation as well as excellent chemotherapy of liposomal PTX are achieved [21-24].
However, the relationship between the structure of conjugated lipids and drug release, pharmacokinetics, tumor accumulation, and anti-tumor activity remains unclear. It is anticipated that changes in chain length of lipids could impact the in vivo behaviors of liposomal PTX, owing to the steric hindrance effect. Our previous study confirmed that conjugating palmitic acid to PTX significantly increased the anti-tumor efficiency of liposomal PTX in mice [25]. Here, we attempt to conjugate three fatty acids with slight difference in chain lengths (e.g., 14C, 16C and 18C), namely myristic acid (MA), palmitic acid (PA) and stearic acid (SA) to PTX, formulate three PTX prodrugs in liposomes, and further explore their effects on the behaviors of liposomal PTX in vitro and in vivo. Noteworthily, all animal experiments were performed strictly according to the procedures approved by the Laboratory Animal Science Department of Fudan University (Shanghai, China).
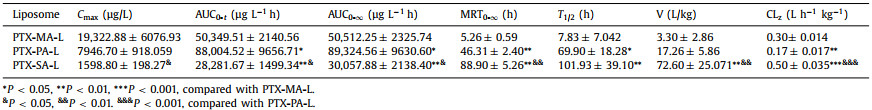
Three PTX prodrugs, namely PTX-MA, PTX-PA and PTX-SA, were designed and synthesized by conjugating MA, PA and SA to PTX via an ester bond, respectively, according to the synthetic routes as shown in Fig. 1A. Their chemical structures were further confirmed by mass spectrometry (MS), 1H nuclear magnetic resonance spectroscopy (NMR) as well as 13C NMR (Fig. S1 in Supporting information).

|
Download:
|
| Fig. 1. (A) Synthesis routes of three PTX prodrugs. (B) Schematic illustration of three prodrug-loaded liposomes. | |
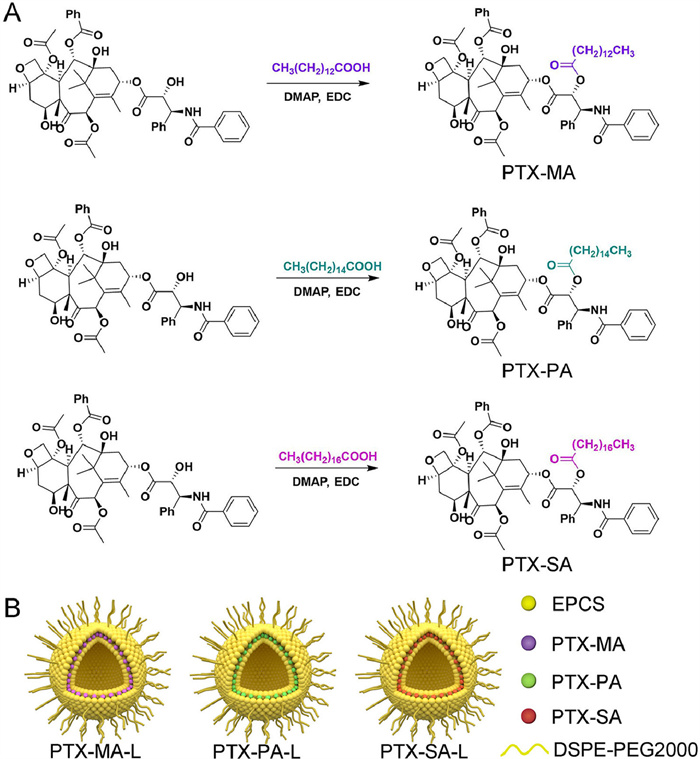
Thin-film hydration followed by extrusion method was utilized to prepare PTX-MA-, PTX-PA- and PTX-SA-loaded liposomes (PTX-MA-L, PTX-PA-L and PTX-SA-L). The obtained liposomes show no precipitation, flocculation, and stratification but uniform dispersion and good fluidity. Cryo-transmission electron microscope (Cryo-TEM) images indicate a spherical shape for all three kinds of liposomes (Figs. 2A–C). The Z-average particle sizes of PTX-MA-L, PTX-PA-L and PTX-SA-L are all around 70 nm, without significant difference (Fig. 2D). We anticipated that such particle size could confer long circulation as well as high accumulation of liposomes in tumors [26], as abundant studies have shown that small particles (< 200 nm) can easily evade the clearance by the mononuclear phagocyte system and penetrate through the interstitial spaces of the leaky vasculatures in tumor regions [27-29].

|
Download:
|
| Fig. 2. Cryo-TEM images of PTX-MA-L (A), PTX-PA-L (B), and PTX-SA-L (C). (D) Distribution profiles of particle sizes of PTX-MA-L, PTX-PA-L and PTX-MA-L. (E) Serum stability of PTX-MA-L, PTX-PA-L and PTX-SA-L. (F) Prodrug release profiles of PTX-MA-L, PTX-PA-L and PTX-SA-L within 24 h. Data expressed as mean ± standard deviation (SD) (n = 3). | |
The polydispersity indexes (PDIs) of these liposomes are all below 0.2, demonstrating a uniform size distribution (Table S1 in Supporting information). The surfaces of three liposomes are weakly negatively charged as their zeta potentials range from −15 mV to −10 mV, which are believed to improve blood compatibility and prolong the circulation of liposomes [30,31]. Thanks to their high lipophilicity, the encapsulation efficiencies (EE%) of PTX-MA, PTX-PA and PTX-SA in liposomes are all over 95% (Table S1). The drug loadings of PTX in PTX-MA-L, PTX-PA-L and PTX-SA-L are all around 7.4%, significantly higher than the reported values of 1.4%–3.7% for PTX liposomes [20,25].
Stability studies show that three liposomes exert excellent stability in plasma. As shown in Fig. 2E and Fig. S2 (Supporting information), all liposomal formulations remain homogeneous in plasma and their particle sizes and PDIs are well maintained within 24 h under incubation at 37 ℃. In addition, all liposomal formulations also demonstrate good storage stability as their particle sizes, PDIs, zeta potentials and EE% are well maintained within two weeks under room temperature (Table S2 in Supporting information). The excellent stability of these liposomes is mainly ascribed to the high compatibility between prodrugs and liposomal components and moreover slightly negatively charged surfaces as well as PEGylation, which can help prevent the aggregation of liposomes by electrostatic repulsion and steric hindrance.
The release profiles of PTX-MA, PTX-PA and PTX-SA from PTX-MA-L, PTX-PA-L and PTX-SA-L are displayed in Fig. 2F. Under sink conditions, PTX-MA-L shows the fastest drug release while PTX-SA-L exhibits the slowest release, indicating a negative correlation between fatty acid chain length and drug release rate [32]. Interestingly, all liposomal formulations show incomplete drug release. Up to 24 h, only 33.4%, 51.7% and 62.0% of prodrugs were released from PTX-SA-L, PTX-PA-L and PTX-MA-L, respectively.
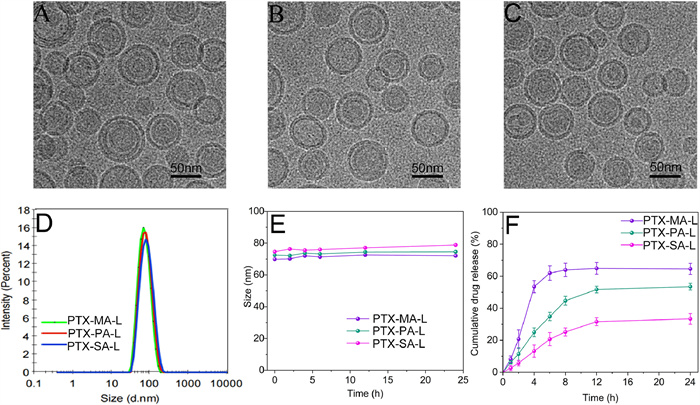
We also investigated in vitro conversion of various prodrugs into PTX using mouse plasma as media, respectively. Fig. 3A shows the profiles of conversion efficiency of PTX-MA, PTX-PA and PTX-SA in mouse plasma versus time. PTX-MA displays the fastest conversion upon incubation in plasma, while PTX-SA exhibits the slowest. This accords with the well acknowledged fact that enzymatic conversion rate of lipid prodrugs to protype drugs is negatively correlated with the chain length of conjugated fatty acids [33]. Besides, fatty acid conjugation increased lipophilicity of PTX, which allows for a long retention of PTX in liposomal carriers. As shown in release studies, PTX-SA has the longest fatty acid chain length among three prodrugs, and consequently, PTX-SA-L exerts the slowest and least prodrug release followed by enzymatic conversion. Noteworthily, all prodrugs show slow conversion rates when encapsulated in liposomes, compared with free ones (Fig. 3B). This is probably attributed to the steric hindrance effect of liposomal carriers, which impedes the direct contact between esterases and prodrugs. Interestingly, all prodrugs display slower conversion rates in rabbit plasma than in mouse plasma, irrespective of whether in a liposomal or free form (Fig. S3 in Supporting information). This indicates that distinct activity and concentration of esterases in various species may impact the in vivo conversion as well as pharmacokinetic parameters of prodrugs.

|
Download:
|
| Fig. 3. (A, B) Conversion profiles of liposomal PTX-MA (PTX-MA-L), PTX-PA (PTX-PA-L), and PTX-SA (PTX-SA-L) in mouse plasma. (C) 4T1 cell migration rate induced by different formulations. (D) 4T1 cell apoptosis rate induced by different formulations. (E–H) Flow cytometry analysis of 4T1 cell apoptosis rate induced by control, PTX-MA-L, PTX-PA-L, and PTX-SA-L. P < 0.05, **P < 0.01, ***P < 0.001. Data expressed as mean ± SD (n = 3). | |
To investigate the interaction between esterases and various prodrugs, molecular docking simulation was conducted using carboxylesterases (CESs) as model esterase. As important phase I metabolizing enzymes, CESs play a crucial role in various pathophysiological processes, such as lipid homeostasis, drug metabolism, bio-detoxification, and are generally believed to predominate in hydrolysis of carboxylesters [34,35]. The results of molecular docking of the CESs with three prodrugs are displayed using Pymol 2.1 software (Fig. S4 in Supporting information). Molecular simulation identifies strong binding affinities between CES1 target proteins and three prodrugs, indicating that a majority of prodrugs are probably hydrolyzed into PTX via CESs.
To predict the anti-tumor capacity of liposomes in vitro, the migration of cancer cells was evaluated as it is closely related to tumor progression. After 24 h exposure, the migration rates in PTX-MA-L, PTX-PA-L and PTX-SA-L groups are 20.44%, 37.58% and 55.23%, respectively, significantly lower than the control group (Fig. 3C and Fig. S5 in Supporting information). Among all prodrug liposomes, PTX-MA-L shows the most potent inhibitory effect on cell migration, while PTX-SA-L exhibits the least inhibition.
Consistent with migration studies, apoptosis assays and cytotoxicity assay indicate that three prodrug liposomes show cytotoxic effects on 4T1 cells compared with the control group, following a descending order of PTX-MA-L > PTX-PA-L > PTX-SA-L (Fig. 3D and Fig. S6 in Supporting information), and the cytotoxicity was dose-dependent.
Fig. S7A (Supporting information) shows that three prodrugs can prevent 4T1 cells from completing G2/M phase, which accords with the action mechanism of PTX that PTX enhances microtubule polymerization to form stable microtubules and therefore arrests tumor cells in the G2/M phase [7]. After treatment by PTX-MA-L, PTX-PA-L and PTX-SA-L, the percentage of 4T1 cells arrested in G2/M phase increased to 60.26%, 42.52% and 16.98%, respectively, significantly higher than the control group (Fig. S7B in Supporting information).
The superior cytotoxicity of PTX-MA-L over PTX-PA-L and PTX-SA-L is attributed to rapid release and rapid conversion of inert PTX-MA to active PTX in tumor cells. PTX-MA has the shortest fatty acid chain length and therefore the fastest conversion rate. After uptake by tumor cells, the cellular concentration of the protype drug, PTX, is the highest in the PTX-MA group while the lowest in the PTX-SA group, which is in accordance with the in vitro cytotoxicity test (Fig. S8 in Supporting information).
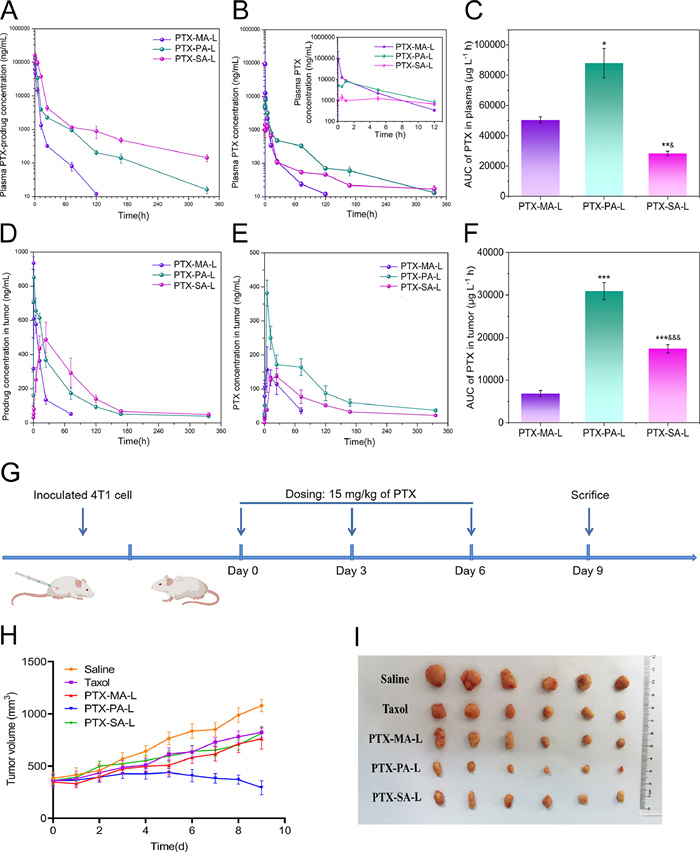
The pharmacokinetics was investigated in mice by simultaneous monitoring of prodrugs and PTX after intravenously administration of three liposomes, namely PTX-MA-L, PTX-PA-L and PTX-SA-L to mice. Fig. 4A shows the pharmacokinetic profiles of PTX-MA, PTX-PA and PTX-SA, while Fig. 4B shows the pharmacokinetic profiles of the corresponding PTX. The corresponding pharmacokinetic parameters obtained by fitting to a non-compartment model are shown in Table 1 and Table S3 (Supporting information), respectively. Among three groups, the plasma concentration and area under the curve (AUC) of PTA-SA were found to be the highest, with those of PTX-MA being the lowest. This complies with the results of drug release studies as well as in vitro conversion assays. The slowest drug release and lipolysis of PTX-SA-L confer PTX-SA the slowest clearance and thereby the longest circulation (Table S3). In contrast, low but lasting PTX concentrations in plasma during the initial 5 h are displayed in the PTX-SA-L group (Fig. 4B), which is probably ascribed to the difficulty in the hydrolysis of PTX-SA in vivo. PTX-MA-L shows the highest plasma PTX concentration within the initial 1 h, which however declines rapidly in the following period owing to the fast hydrolysis of PTX-MA. Distinct from PTX-SA-L and PTX-MA-L, the plasma PTX concentration of PTX-PA-L is initially moderate prior to a peak at 1 h and decreases gradually in the following days.

|
Download:
|
| Fig. 4. (A, B) Pharmacokinetic profiles of three PTX-prodrugs (A) and corresponding PTX (B) after administration of various liposomes to mice (n = 3). The inset shows the corresponding enlargement of the profile (B) within the first 12 h. (C) AUC values of plasma PTX after administration of various liposomes to mice (n = 3). (D, E) Plotting the PTX-prodrugs (D) and the corresponding PTX (E) concentrations in tumors versus time after administration of various liposomes to mice (n = 3). (F) AUC values of PTX concentration in tumors after treatment with PTX-MA-L, PTX-PA-L and PTX-SA-L (n = 3). (G) Schematic time diagram of animal grouping and dosing. (H) Plotting the tumor volume versus time after treatment with various formulations (n = 6). (I) Representative images of solid tumors harvested on day 9. P < 0.05, **P < 0.01, ***P < 0.001, compared with PTX-MA-L; &P < 0.05, &&P < 0.01, &&&P < 0.001, compared with PTX-PA-L. Data expressed as mean ± SD. | |
|
|
Table 1 Pharmacokinetic parameters of PTX converted after intravenous administration of PTX-MA-L, PTX-PA-L and PTX-SA-L to mice, respectively (n = 3). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The clearance rates (CLzs) of PTX-MA, PTX-PA and PTX-SA are 0.067 ± 0.011, 0.029 ± 0.0015 and 0.013 ± 0.0023 L h−1 kg−1, respectively, significantly lower than those of corresponding converted PTX (0.30 ± 0.014, 0.17 ± 0.017 and 0.50 ± 0.035 L h−1 kg−1, respectively). And the CLzs of prodrugs are positively correlated with the enzymatic hydrolysis as well as drug release, which indicates that prodrugs are mainly cleared in the form of PTX. Therefore, a moderate conversion rate of PTX-PA to PTX can continuously compensate for the rapid elimination of PTX and impart a relatively high PTX concentration in plasma, which finally endows PTX-PA-L with the highest AUC of PTX than the others (Fig. 4C). This is interesting as neither rapid degradation nor slow conversion of prodrugs favors drug retention in circulation.
The accumulation of lipid-PTX prodrugs as well as PTX in tumors was also simultaneously investigated in mice after intravenous administration of PTX-MA-L, PTX-PA-L and PTX-SA-L. As shown in Figs. 4D and E, the Tmax values for PTX-MA, PTX-PA and PTX-SA accumulation in tumors are 0.5, 1 and 24 h, respectively, which are much smaller than those of corresponding PTX. The slow drug release of PTX-SA from liposomes probably contributes to its large Tmax value, while its slow conversion rate enables a large Tmax value of corresponding PTX in tumors.
Consistent with the pharmacokinetic results, PTX-PA-L shows moderate accumulation of PTX-PA, but the highest concentration as well as AUC of PTX in tumors among all liposomal formulations (Figs. 4D–F). This indicates that higher AUC of plasma PTX favors tumor accumulation, probably owing to the fact that distribution of released PTX into tumors partly contributed to the total PTX accumulated in tumors. Noteworthily, lipid conjugation may also influence the tumor accumulation of liposomes, which needs further verification by monitoring of intact liposomes. Labeling by an aACQ probe can help to accurately identify and semi-quantify liposomes in tumors [36-39]. Over a long period, the tumoral PTX concentration in PTX-PA-L group is much higher than the other two groups, indicating that PTX-PA-L probably exerts a better anti-tumor efficiency in vivo.
The in vivo anti-tumor efficiencies of three PTX prodrug liposomes were evaluated in 4T1 tumor-bearing mice, using Taxol® as a control (Fig. 4G). After administration of various formulations on days 0, 3 and 6, PTX-PA-L shows significantly higher anti-tumor efficacy than the other two liposomes as well as Taxol®, while PTX-MA-L and PTX-SA-L show comparable effect as Taxol® (Figs. 4H and I). This is mainly ascribed to the fact that PTX-PA-L has the highest delivery efficiency of PTX to tumors, as discussed above. The tumor inhibition rates of PTX-MA-L, PTX-PA-L, PTX-SA-L and Taxol are calculated to be 54.99%, 78.44%, 51.80% and 55.39%, respectively (Fig. S9A in Supporting information). By H & E staining and TUNEL staining, the highest necrosis as well as apoptosis rate was observed in the PTX-PA-L group, confirming the strong anti-tumor efficacy of PTX-PA-L (Fig. S10 in Supporting information).
Noteworthily, all liposomal formulations did not result in any significant change in the body weight of mice throughout the experimental period. By hematological analysis, the mean blood cell levels in all groups were evaluated. As shown in Table S4 (Supporting information), Taxol® induces a significant decrease in white blood cells (WBC) and neutrophils (NEUT), indicating its high myelosuppression efficiency, while the WBC and NEUT counts after administration of PTX-PA-L or PTX-SA-L are significantly increased compared with Taxol®. This is probably ascribed to the low plasma PTX concentration contributed by slow drug release and conversion of PTX-PA-L and PTX-SA-L, as well as high biocompatibility of liposomal components. In contrast, PTX-MA-L treatment results in slightly increased WBC and NEUT counts compared with Taxol®, whereas the differences are not significant probably owing to its rapid drug release and conversion. Other indexes, such as red blood cells (RBC), platelet (PLT) and hemoglobin (HGB) counts, are also significantly improved for PTX-PA-L and PTX-SA-L compared with Taxol®, which double verifies the excellent safety of PTX-PA-L and PTX-SA-L.
In conclusion, we designed and successfully synthesized a series of lipid-PTX prodrugs by conjugating fatty acids with various chain lengths, namely MA, PA and SA, to PTX via an ester bond, respectively. Owing to their excellent lipophilicity, three prodrugs can be stably incorporated into liposomes with high encapsulation efficiencies and drug loadings. These prodrug-loaded liposomes show tunable prodrug release and in vitro conversion of prodrugs into PTX, which are negatively correlated with the chain length of fatty acids. A shorter chain length favors faster drug release as well as enzymatic conversion to active PTX, which enables higher in vitro anti-tumor capacity.
In addition to drug release and conversion, the minor changes in chain lengths of conjugated lipids can also significantly impact the in vivo behaviors of the constructed liposomal drug delivery systems, such as pharmacokinetics, tumor accumulation and in vivo anti-tumor efficiency. A long chain length (18C) confers a slow clearance as well as long circulation of PTX-SA, but a low plasma concentration and AUC of PTX owing to its difficulty in drug release and conversion. Consequently, PTX-SA-L has the highest AUC of PTX-SA but the slowest accumulation of PTX in tumors. In contrast, a short chain length (14C) enables a rapid blood clearance of PTX-MA as well as PTX, and a fast but low accumulation of PTX-MA as well as PTX. Compared with MA and SA, the moderate chain length of PA endows PTX-PA with moderate drug release, conversion rate, blood circulation and tumor accumulation. However, the fit drug release and enzymatic conversion coincidently satisfy continuous compensation for the rapid elimination of PTX and support a long circulation and tumor accumulation of PTX.
With the highest accumulation of PTX in tumors, PTX-PA-L shows the most potent anti-tumor capacity in vivo compared with the other two liposomes. Witnessing its excellent safety, PTX-PA-L is believed to hold potential for clinical translation.
Our results show that slight difference in the chain length of conjugated lipid has a non-negligible impact on the pharmacokinetics and anti-tumor efficiencies of prodrug liposomes. Neither long chain length nor short one facilitates a long circulation as well as superior anti-tumor efficacy of chemotherapeutic agents. The structure of conjugated lipids should be taken into careful consideration upon design and optimization of prodrug formulations. It is necessary to comprehensively consider the compatibility between prodrugs and nanocarriers, in vivo drug release and enzymatic conversion rate. Fatty acids with appropriate chain length should be selected to confer a proper drug release, and enhanced tumor accumulation. Drug delivery efficiency of protype drug to tumors seems to predominate cell uptake efficiency in anti-tumor efficiency. This indicates that in vitro cytotoxicity cannot accurately predict in vivo behaviors. Merely increasing the tumor accumulation or cell uptake of prodrugs does not guarantee an excellent in vivo anti-tumor capacity, as most prodrugs are bioinert. However, introducing stimuli-cleavable linkages may circumvent this dilemma.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (Nos. 82273867, 82030107), Shanghai Science and Technology Project of Little Giant (No. 1902HX76600), Shanghai Qingpu District Industry-University-Research Cooperative Development Funding Project (No. 2022–7), High-level Talents of Fujian University of Chinese Medicine (No. X2019006-Talents).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108756.
| [1] |
H. Sung, J. Ferlay, R.L. Siegel, et al., CA Cancer J. Clin. 71 (2021) 209-249. DOI:10.3322/caac.21660 |
| [2] |
M. Arnold, E. Morgan, H. Rumgay, et al., Breast 66 (2022) 15-23. DOI:10.1016/j.breast.2022.08.010 |
| [3] |
H.S. Wang, Y.Y.Gao L.Wang, et al., Chin. Chem. Lett. 32 (2021) 1041-1045. DOI:10.1016/j.cclet.2020.08.044 |
| [4] |
Y. Wang, J. Yu, D. Li, et al., J. Control. Release 341 (2022) 812-827. DOI:10.1016/j.jconrel.2021.12.023 |
| [5] |
A.M. Sofias, M. Dunne, G. Storm, C. Allen, Adv. Drug Deliv. Rev. 122 (2017) 20-30. DOI:10.1016/j.addr.2017.02.003 |
| [6] |
W.J. Gradishar, S. Tjulandin, N. Davidson, et al., J. Clin. Oncol. 23 (2005) 7794-7803. DOI:10.1200/JCO.2005.04.937 |
| [7] |
S.C. Kim, D.W. Kim, Y.H. Shim, et al., J. Control. Release 72 (2001) 191-202. DOI:10.5223/kjpgn.2001.4.2.191 |
| [8] |
J.A. Jackman, T. Meszaros, T. Fulop, et al., Nanomedicine 12 (2016) 933-943. DOI:10.1016/j.nano.2015.12.377 |
| [9] |
Q.Q. Xiao, X.T. Li, C. Liu, et al., Chin. Chem. Lett. 33 (2022) 4191-4196. DOI:10.1016/j.cclet.2022.01.083 |
| [10] |
N. Yin, W.J. Zhang, R.X. Wei, et al., Asian J. Pharm. Sci. 17 (2022) 867-879. DOI:10.1016/j.ajps.2022.10.001 |
| [11] |
G.F. Boafo, Y.J. Shi, Q.Q. Xiao, et al., Chin. Chem. Lett. 33 (2022) 4600-4604. DOI:10.1016/j.cclet.2022.04.033 |
| [12] |
T. Takayama, T. Shimizu, A.S.A. Lila, et al., Pharmaceutics 12 (2020) 990. DOI:10.3390/pharmaceutics12100990 |
| [13] |
N. Tian, D. Wang, X. Li, M. Xue, B. Zheng, Evid. Based Complement. Alternat. Med. 30 (2022) 5651793. DOI:10.2478/s13380-013-0126-1 |
| [14] |
L. Ye, J. He, Z. Hu, et al., Food Chem. Toxicol. 52 (2013) 200-206. DOI:10.1016/j.fct.2012.11.004 |
| [15] |
J. Zhang, Y. Pan, Q. Shi, et al., Cancer Commun. 42 (2022) 3-16. DOI:10.1002/cac2.12225 |
| [16] |
H. Zhou, J. Yan, W. Chen, et al., Front. Oncol. 10 (2021) 1731. DOI:10.3389/fonc.2020.01731 |
| [17] |
H. Chen, S. Huang, H. Wang, et al., Drug Deliv. 28 (2021) 1067-1079. DOI:10.1080/10717544.2021.1921078 |
| [18] |
B. Ahmad, F. Shireen, A. Rauf, et al., IET Nanobiotechnol. 15 (2021) 1-18. DOI:10.1049/nbt2.12007 |
| [19] |
H. Xu, W. Cui, Z. Zong, et al., Drug Deliv. 29 (2022) 2414-2427. DOI:10.1080/10717544.2022.2103209 |
| [20] |
J.A. Zhang, G. Anyarambhatla, L. Ma, et al., Eur. J. Pharm. Biopharm. 59 (2005) 177-187. DOI:10.1016/j.ejpb.2004.06.009 |
| [21] |
R. Du, T. Zhong, W.Q. Zhang, et al., Int. J. Nanomed. 9 (2014) 3091-3105. DOI:10.2147/IJN.S65664 |
| [22] |
S. Mura, D.T. Bui, P. Couvreur, J. Nicolas, J. Control. Release 208 (2015) 25-41. DOI:10.1016/j.jconrel.2015.01.021 |
| [23] |
L. Ling, Y. Du, M. Ismail, et al., Int. J. Pharm. 526 (2017) 11-22. DOI:10.1016/j.ijpharm.2017.04.024 |
| [24] |
Z. Wang, L. Ling, Y. Du, C. Yao, X. Li, Int. J. Pharm. 564 (2019) 244-255. DOI:10.3390/fi11110244 |
| [25] |
D. Cheng, N. Yu, Y.F. Xu, et al., Chin. Pharmaceut. J. 53 (2018) 614-619. |
| [26] |
F. Ravar, E. Saadat, M. Gholami, et al., J. Control. Release 229 (2016) 10-22. DOI:10.1016/j.jconrel.2016.03.012 |
| [27] |
S. Maritim, P. Boulas, Y. Lin, Int. J. Pharm. 592 (2021) 120051. DOI:10.1016/j.ijpharm.2020.120051 |
| [28] |
C. Bi, X.Q. Miao, S.F. Chow, et al., Nanomedicine 13 (2017) 943-953. DOI:10.1016/j.nano.2016.11.004 |
| [29] |
K.S. Chu, W. Hasan, S. Rawal, et al., Nanomedicine 9 (2013) 686-693. DOI:10.1016/j.nano.2012.11.008 |
| [30] |
Y. Wu, B. Mou, S. Song, et al., Food Res. Int. 136 (2020) 109301. DOI:10.1016/j.foodres.2020.109301 |
| [31] |
S. Huang, X. Wang, M. Liu, et al., Drug Deliv. 29 (2022) 1132-1141. DOI:10.1080/10717544.2022.2058114 |
| [32] |
T. Gaekens, M. Guillaume, H. Borghys, et al., J. Control. Release 232 (2016) 196-202. DOI:10.1016/j.jconrel.2016.04.029 |
| [33] |
Y. Ma, H. He, F. Xia, et al., Nanomedicine 13 (2017) 2643-2654. DOI:10.1016/j.nano.2017.07.014 |
| [34] |
M.J. Hatfield, R.A. Umans, J.L. Hyatt, et al., Chem. Biol. Interact. 259 (2016) 327-331. DOI:10.1016/j.cbi.2016.02.011 |
| [35] |
T. Satoh, M. Hosokawa, Chem. Biol. Interact. 162 (2006) 195-211. DOI:10.1016/j.cbi.2006.07.001 |
| [36] |
H. He, L. Wang, Y. Ma, et al., J. Control. Release 327 (2020) 725-736. DOI:10.1016/j.jconrel.2020.09.024 |
| [37] |
Y. Cai, J. Qi, Y. Lu, et al., Adv. Drug Deliv. Rev. 188 (2022) 114463. DOI:10.1016/j.addr.2022.114463 |
| [38] |
H. He, C. Liu, J. Ming, et al., Aggregate 3 (2022) e163. DOI:10.1002/agt2.163 |
| [39] |
Y. Cai, X. Ji, Y. Zhang, et al., Aggregate 4 (2023) e277. DOI:10.1002/agt2.277 |