 2024, Vol. 35
2024, Vol. 35 


b Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, China
Cyclic polymers have unique properties compared with their linear analogues due to their “endless” topology, such as smaller radius of gyration and hydrodynamic volume, lower viscosity, and higher glass transition temperature [1–4]. Given that, cyclic polymers have been successfully applied to numerous fields including gene transfection [5,6], drug delivery [7,8], surface modification [9,10] and so on [11–14]. Currently, two main methods can be utilized to synthesize cyclic polymers: ring-closure [15] and ring-expansion methods [16,17]. Ring-expansion method can be used to generate cyclic polymers by inserting cyclic monomers into a cyclic catalyst/initiator repeatedly. Cyclic polymers with high molecular weights (> 1 × 106 g/mol) can be obtained at high monomer concentrations by ring-expansion method [18–20]. However, this method often lacks control over molecular weights and polydispersities of cyclic polymers [21]. Meanwhile, the formidable synthetic challenge of cyclic catalysts and specific monomers limit its extensive application [22]. Ring-closure method can be applied to construct cyclic polymers by cyclizing a linear polymer via an end-group coupling reaction. Combined that with living polymerization, cyclic polymers with controlled molecular weights and narrow polydispersities can be prepared precisely [23,24]. Nevertheless, in order to suppress the intermolecular coupling reaction, ring-closure method is usually conducted under highly dilute conditions (< 10-5 mol/L) [15]. Simultaneously, according to Jacobson–Stockmayer theory, the probability of cyclization reaction decreases with the increase of chain length of linear polymers [25]. Thus, access to high-molecular-weight (> 25,000 g/mol) cyclic polymers at high concentrations by ring-closure method is still a challenge.
In nature, biomacromolecules such as DNA, proteins and enzymes can form a high-level structure by the self-folding process. Recently, single chain folding technique has been employed to create single chain polymeric nanoparticles (SCPNs) in water from the amphiphilic random copolymers with hydrophobic and hydrophilic pendants [26–29]. For example, Sawamoto et al. applied random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) and hydrophobic alkyl pendants to form unimer micelles carrying hydrophobic cores covered by hydrophilic pendants [30,31]. It was found that the self-folding process was influenced by the content of hydrophilic units in polymer chains. The amphiphilic copolymers tended to form SCPNs instead of multi-chain micelles with the increase of the hydrophilic unit contents in copolymers. Therefore, by adjusting the content of the hydrophilic units, pure SCPNs can be efficiently obtained.
The self-folding process can shorten the distance between the ends of the same polymer chain and significantly prevent intermolecular reactions, thus the entropic penalty of end-group coupling induced by long polymer chain can be reduced. Recently, we proposed a unique self-folding cyclization technique to produce well-defined cyclic polymers. The technique involves the use of linear P(OEGA-co-DDA) precursors with a molar ratio of 7:3 between oligo(ethylene glycol) acrylate (OEGA) and dodecyl acrylate (DDA) units [32]. Cyclic polymers with high molecular weights (> 105 g/mol) were prepared at relatively high concentrations (40 mg/mL) in aqueous solution by this approach.
Currently, one of the most critical issues for the large-scale preparation of cyclic polymers by ring-closure method is how to prepare pure cyclic polymers at high concentrations. Most ring-closure strategies need to be performed at low concentrations, which greatly raises the production costs of cyclic polymers. Based on our previous work [32], we wonder if the self-folding cyclization technique can be used to prepare cyclic polymers at higher concentrations by adjusting the ratios of hydrophilic and hydrophobic units in linear amphiphilic random copolymers. As shown in Fig. 1, linear P(OEGA-co-DDA) precursors with two anthryl end groups and different contents of OEGA units were prepared by RAFT polymerization. Subsequently, linear P(OEGA-co-DDA) copolymers underwent the self-folding process to generate SCPNs. By irradiating the SCPNs with UV light (365 nm), cyclization was achieved via a Diels–Alder (D–A) reaction between anthryl end groups, and cyclic polymers can be obtained efficiently without a tedious purification procedure. We investigated the effects of the polymer concentrations and the ratios of hydrophilic and hydrophobic moieties on the purity of the obtained cyclic polymers, and prepared cyclic polymers with high purity at high polymer concentrations (up to 90 mg/mL) by adjusting the ratios of the hydrophilic and hydrophobic units in linear precursors. This approach is of great significance for large-batch production of well-defined cyclic polymers with high molecular weights (> 105 g/mol).

|
Download:
|
| Fig. 1. Schematic illustration of the preparation of cyclic P(OEGA-co-DDA) copolymers at high concentrations. (a) Synthetic route of cyclic P(OEGA-co-DDA) copolymers via self-folding cyclization technique. (b) Illustration of the effect of the ratios of OEGA and DDA units in linear precursors on the purity of cyclic polymers prepared by self-folding cyclization technique. | |
In order to investigate the effects of the hydrophilic unit contents on the purity of the obtained cyclic polymers, linear random copolymers poly((oligo(ethylene glycol) acrylate)-co-(dodecyl acrylate)) (P(OEGA-co-DDA)) were prepared. A bifunctional chain transfer agent (BCTA) with two anthryl groups was synthesized (Scheme S1 in Supporting information), and its structure was confirmed by the 1H and 13C NMR spectra in Figs. S1 and S2 (Supporting information). Subsequently, linear P(OEGA-co-DDA) copolymers with different molar ratios of [OEGA]/[DDA] were synthesized via RAFT copolymerization of OEGA (Mn = 482 g/mol) and DDA in toluene (the feed molar ratios of [OEGA]/[DDA]/[BCTA] were set as 750/250/1,800/200/1,900/100/1, and the corresponding linear copolymers were denoted as L1, L2 and L3, respectively). The 1H NMR spectra of the resultant copolymers are shown in Fig. S3 (Supporting information), several anthryl proton peaks at 7.4–8.5 ppm (peaks s, r, q) and peaks at approximately 7.2 ppm (peaks a, b) belonging to phenyl protons of the BCTA segment were observed, as well as the characteristic resonances of –OCH of OEGA (peak n) and –CH2CH of DDA (peak h) at 3.4 ppm and 0.8 ppm, respectively. By comparing the integral area of the signal of methyl protons in OEGA (peak n) with that in DDA (peak h), the contents of OEGA units in linear P(OEGA-co-DDA) copolymers were determined to be 72% (L1), 77% (L2) and 88% (L3), respectively. The UV‒vis spectra of linear P(OEGA-co-DDA) copolymers are shown in supporting information (blue curves in Figs. S16-S18 in Supporting information), from which the characteristic absorbance band at 365 nm ascribed to the π–π * transition of the anthryl groups can be observed.
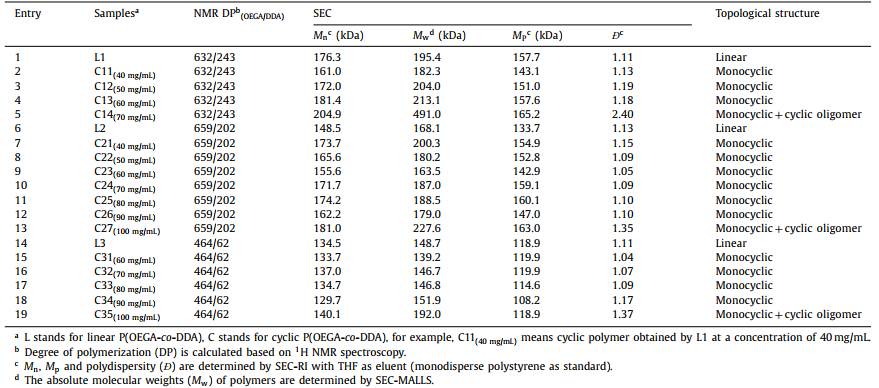
Three-detection size exclusion chromatography (TD-SEC) equipped with multiangle laser light scattering (MALLS) detector was utilized to characterize the molecular weights and polydispersities of P(OEGA-co-DDA) copolymers. As shown in Fig. 2, the SEC-MALLS traces of linear copolymers revealed unimodal peaks with narrow distribution, and the number-average molecular weights (Mn) were 176.3 kDa (L1, Ð = 1.11), 148.5 kDa (L2, Ð = 1.13), 134.5 kDa (L3, Ð = 1.11), respectively. These results indicate that well-defined linear P(OEGA-co-DDA) copolymers with anthryl end groups were synthesized successfully.

|
Download:
|
| Fig. 2. SEC measurements of linear P(OEGA-co-DDA) precursors and cyclic P(OEGA-co-DDA) copolymers using THF as the eluent. The SEC-MALLS traces of L1 (a), L2 (b) and L3 (c) before and after cyclization at different polymer concentrations. | |
According to the previous reports [33,34], amphiphilic copolymers can self-fold in water to form SCPNs derived by the hydrophobic and hydrophilic interaction of the alkyl pendants, polymer backbones and PEG pendants. In this work, we investigated the self-folding behaviors of linear P(OEGA-co-DDA) random copolymers, and the emphasis is the effects of polymer composition and concentration on the resulting polymeric nanoparticles in water. L1, L2 and L3 were dissolved in pure water at different concentrations. The self-folding process of P(OEGA-co-DDA) copolymers (L1-L3) in water was investigated with 1H NMR, TEM and DLS. Figs. S4-S6 (Supporting information) are the 1H NMR spectra of L1-L3 in D2O at room temperature, in which the proton signals of hydrophobic backbone (peaks d, e, i, j) and dodecyl pendants (peaks g, h) became broader and the integral ratio of peak “h” relative to peak “n” decreased obviously in D2O compared with that in CDCl3, indicating the occurrence of self-folding [34].
In addition, TEM was utilized to investigate the morphologies of the polymeric nanoparticles prepared via self-folding, and the TEM images are shown in Figs. S7-S9 (Supporting information). NP-L1–60 (the polymeric nanoparticles obtained via self-folding of L1 at a concentration of 60 mg/mL) (Fig. S7a), NP-L2–60 (Fig. S8a) and NP-L3–60 (Fig. S9a) were small spherical micelles with diameters of 5-10 nm, which corresponded to the size of the unimer micelles reported by Sawamoto's group [31], indicating the formation of SCPNs for all samples at a concentration of 60 mg/mL. When the concentration of the polymers further increased, larger nanoparticles can be observed. For example, TEM images of NP-L1–70 (Fig. S7b), NP-L2–100 (Fig. S8b) and NP-L3–100 (Fig. S9b) showed the existence of nanoparticles with diameters of approximately 10-30 nm, possibly because of the formation of multichain nanoparticles. The TEM results reveal that L2 and L3 which contained higher hydrophilic unit contents compared to L1 produced multichain nanoparticles by self-folding at higher concentrations.
Subsequently, the nanoparticles obtained at different polymer concentrations were analyzed by DLS measurement, and the results are given in Figs. S10-S12 (Supporting information). NP-L1–60, NP-L2–60 and NP-L3–60 had small hydrodynamic diameters (Dh: ~10 nm) (blue curves in Figs. S10a, S11a and S12a), though NP-L1–70, NP-L2–100 and NP-L3–100 had larger hydrodynamic diameters (Dh: 20-40 nm) because of the presence of multichain nanoparticles (blue curves in Figs. S10b, S11b and S12b). These results demonstrate that the composition and concentration of the linear amphiphilic random copolymers had significant impacts on the self-folding behavior. On the other hand, by controlling the molar ratios of the OEGA and DDA units and the concentrations of copolymers, SCPNs can be successfully obtained by the self-folding process.
After self-folding, the cyclization process was performed by irradiating the aqueous solution of the nanoparticles generated by L1, L2 and L3 with 365 nm UV light for 4 h. Cyclic P(OEGA-co-DDA) copolymers were obtained by freeze-drying. As shown in the 1H NMR spectra of the resultant copolymers (Figs. S13-S15 in Supporting information), the typical signals of anthryl protons at 7.4–8.5 ppm almost disappeared. Meanwhile, new signals at 6.8 ppm (peaks q′, r′), and 5.5 ppm (peak s′) of the anthracene dimer were observed, indicating the occurrence of photo-dimerization reaction of anthryl groups. UV‒vis spectroscopy was also utilized to investigate the light-triggered dimerization reaction between anthryl groups, and the results are shown in Figs. S16-S18. Compared to the UV‒vis spectra of linear P(OEGA-co-DDA) copolymers, the characteristic absorbance band of the anthryl group at 365 nm disappeared after irradiation. These results demonstrated that the anthryl end groups of linear precursors were transformed into anthracene dimers. The sizes of the nanoparticles after irradiation were analyzed by DLS (Figs. S10-S12), and no significant changes were observed compared to those before irradiation, which indicated that the light-triggered dimerization reaction occurred mainly within nanoparticles rather than between nanoparticles.
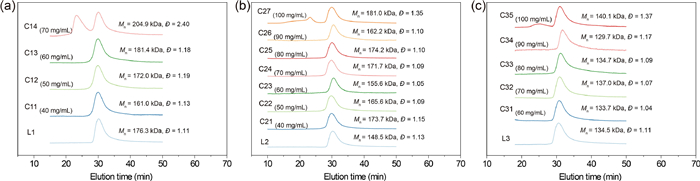
To further investigate the cyclic topology and purity of the resultant polymers, TD-SEC analysis and TEM were employed. Considering that cyclic polymer has a smaller hydrodynamic volume than its linear counterpart, the SEC equipped with differential refractive index detector (SEC-RI) is usually used to characterize cyclic polymers. Generally, the SEC-RI trace of polymer shifts to the lower molecular weight direction after cyclization [35–37]. However, in the current work, the peak shift of the cyclic polymers was imperceptible, probably because that the long side chains connected to the polymer backbone resulted in little conformational difference between the cyclic polymer and its linear precursor [38]. The summary of the molecular weight data of linear and cyclic polymers is listed in Table 1. All samples revealed monomodal evolution of SEC-RI traces (Figs. S19-S21 in Supporting information) with relatively low molecular weight distributions. Taking into account that the MALLS detector is more sensitive to the high molecular weight fraction than the differential refractive index detector [39], SEC-MALLS was further employed to investigate the purity of cyclic polymers. As shown in Fig. 2, the SEC-MALLS traces of the resultant cyclic polymers obtained from L1 at concentrations of 40 mg/mL (ca. 2.3 × 10−4 mol/L for L1) (the corresponding polymer was denoted as C11), 50 mg/mL (ca. 2.8 × 10−4 mol/L for L1) (C12), 60 mg/mL (ca. 3.4 × 10−4 mol/L for L1) (C13) all revealed unimodal peaks with narrow distributions. However, when the concentration of L1 increased to 70 mg/mL (ca. 4.0 × 10−4 mol/L for L1) (C14), a bimodal curve with a broad distribution was observed, which indicated the occurrence of intermolecular reaction due to the presence of multi-chain nanoparticles. The peak at 23.84 min (Peak 1 in Fig. 3a) was attributed to cyclic oligomer (intermolecular reaction product), and the peak at 30.42 min (Peak 2 in Fig. 3a) was attributed to monocyclic polymer (intramolecular reaction product). By analyzing the deconvolution of the SEC-MALLS trace of the products (Fig. 3a), the contents of the monocyclic polymer and cyclic oligomer can be calculated as 56.29% and 43.71%, respectively. For L2 and L3, it should be emphasized that high-purity monocyclic polymers can be obtained even when the polymer concentration was up to 90 mg/mL (ca. 6.1 × 10−4 mol/L for L2, 6.7 × 10−4 mol/L for L3) (the corresponding polymers were denoted as C26 and C34, respectively), which is higher than previously reported polymer concentration in ring-closure method [24,32,40] for preparing monocyclic polymers. When the polymer concentration further increased to 100 mg/mL (ca. 6.7 × 10−4 mol/L for L2, 7.4 × 10−4 mol/L for L3), a shoulder peak appeared in the direction of high molecular weight in the SEC-MALLS traces of cyclic polymers obtained from L2 and L3 (C27 and C35). Horizontally, as estimated by the deconvolution of the SEC-MALLS traces of the products after irradiation (Figs. 3b and c), the contents of monocyclic polymer (Peak 3 in Fig. 3b and Peak 2 in Fig. 3c) were 73.07% for C27 and 89.71% for C35. Higher percentage of monocyclic polymers can be obtained from L3 compared to L2 at a concentration of 100 mg/mL by intramolecular reaction, which was probably because that higher content of hydrophilic units could provide better stabilization of SCPNs in aqueous solution, and higher proportion of SCPNs were able to be generated from L3 than L2 at the same polymer concentration.
|
|
Table 1 Characterization of P(OEGA-co-DDA) copolymers. |
{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. Deconvolution of SEC-MALLS traces for cyclic polymers through the fitting of each peak. (a) Fitting result of deconvoluted peaks of SEC trace for C14. Cumulative curve of C14: blue, Peak 1: black, Peak 2: red. (b) Fitting result of deconvoluted peaks of SEC trace for C27. Cumulative curve of C27: green, Peak 1, Peak 2, Peak 3: blue. (c) Fitting result of deconvoluted peaks of SEC trace for C35. Cumulative curve of C35: orange, Peak 1: green, Peak 2: purple. | |
{kind=link}
Observation of the cyclic topology by high-resolution transmission electron microscopy (HRTEM) would provide more direct evidence for cyclization [41,42]. Generally, visualization of cyclic polymers was achieved by grafting side chains onto the backbone [43,44]. In this work, since the existence of OEGA and DDA pendants greatly stretched the polymer backbone and improved the mass density of the polymer backbone [45,46], the visualization of cyclic P(OEGA-co-DDA) by TEM is promising. C14, C27 and C35 were chosen as representative examples for investigating the visualization of cyclic polymers. As shown in Figs. 4a-c, polymers with monocyclic structures could be observed. The average diameters of these cyclic copolymer backbones were approximately 67 nm for C14, 57 nm for C27, and 42 nm for C35, respectively. Based on the bond length of C–C (0.154 nm) [47], the theoretical diameters corresponding to C14, C27 and C35 were calculated to be 70 nm, 69 nm and 42 nm, respectively. In Figs. 4d-f, cyclic oligomers with diameters of around 100 nm were observed, and the diameters were larger than the theoretical values of monocyclic polymers. It might be caused by intermolecular coupling at high polymer concentrations. This result is consistent with the appearance of multiple peaks in the SEC-MALLS traces of cyclic P(OEGA-co-DDA) copolymers obtained at high polymer concentrations in Fig. 2. The visualization of cyclic polymers by HRTEM directly confirmed the cyclic topology of the resultant polymers. The above results reveal that by adjusting the ratios of the hydrophilic and hydrophobic units in linear precursors, high-purity monocyclic polymers can be successfully produced at higher concentrations up to 90 mg/mL (6.7 × 10−4 mol/L for L3).

|
Download:
|
| Fig. 4. TEM images of cyclic polymers obtained from linear precursors at different polymer concentrations. (a-c) HRTEM images of monocyclic polymers observed from C14, C27 and C35, respectively. TEM images of cyclic polymers (d) C14, (e) C27, (f) C35. Inset: images of cyclic oligomers observed from C14, C27 and C35. | |
{kind=link}
In this investigation, self-folding cyclization technique for preparing cyclic polymers at higher concentrations by adjusting the content of the hydrophilic units in linear copolymer chains has made satisfactory progress. Compared with the traditional ring-closure method, this method allows preparing monocyclic polymers at higher concentrations, but it still falls slightly short in polymer concentration compared with the ring-expansion method. Therefore, we will attempt to further improve the self-folding cyclization technique by molecule design, such as introduction of intramolecular interactions in polymer chain to increase the polymer concentration for obtaining pure monocyclic polymers.
In summary, we investigated the effects of the ratios of hydrophilic and hydrophobic moieties in linear P(OEGA-co-DDA) copolymers and polymer concentration on the purity of the cyclic polymers obtained by the self-folding cyclization technique. By adjusting the ratios of hydrophilic OEGA and hydrophobic DDA units in anthracene-terminated linear precursors, SCPNs can be generated at high polymer concentrations. Monocyclic P(OEGA-co-DDA) copolymers with relatively high molecular weights were produced via irradiating the aqueous solution of the polymeric nanoparticles with UV light. The polymer concentration in the self-folding cyclization technique for preparing high-purity monocyclic polymers can reach as high as 90 mg/mL (6.7 × 10−4 mol/L for L3). This work provides a promising method to achieve large-scale production of cyclic polymers.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe financial support from the National Natural Science Foundation of China (Nos. 22201276, 22131010, 52021002) and the Fundamental Research Funds for the Central Universities (No. WK2060000012) is gratefully acknowledged. The authors thank Prof. B. B. Jiang, Mr. Y. Z. Du and Mr. L. Fang from Changzhou University for their assistance with TD-SEC characterization.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108956.
| [1] |
T. McLeish, Science 297 (2002) 2005-2006. DOI:10.1126/science.1076810 |
| [2] |
M. Kapnistos, M. Lang, D. Vlassopoulos, et al., Nat. Mater. 7 (2008) 997-1002. DOI:10.1038/nmat2292 |
| [3] |
S.M. Grayson, J.M.J. Fréchet, Chem. Rev. 101 (2001) 3819-3868. DOI:10.1021/cr990116h |
| [4] |
Z. Lu, B. Guo, Y. Zhao, L. Hou, L. Xiao, Chin. Chem. Lett. 33 (2022) 825-829. DOI:10.1016/j.cclet.2021.07.033 |
| [5] |
F.P. Seib, A.T. Jones, R. Duncan, J. Control. Release 117 (2007) 291-300. DOI:10.1016/j.jconrel.2006.10.020 |
| [6] |
H. Wei, D.S.H. Chu, J. Zhao, J.A. Pahang, S.H. Pun, ACS Macro Lett. 2 (2013) 1047-1050. DOI:10.1021/mz400560y |
| [7] |
V.P. Torchilin, A.N. Lukyanov, Z. Gao, B. Papahadjopoulos-Sternberg, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 6039-6044. DOI:10.1073/pnas.0931428100 |
| [8] |
X. Wan, T. Liu, S. Liu, Biomacromolecules 12 (2011) 1146-1154. DOI:10.1021/bm101463d |
| [9] |
M.A. Aboudzadeh, A. Iturrospe, A. Arbe, M. Grzelczak, F. Barroso-Bujans, ACS Macro Lett. 9 (2020) 1604-1610. DOI:10.1021/acsmacrolett.0c00730 |
| [10] |
G. Morgese, B. Shirmardi Shaghasemi, V. Causin, et al., Angew. Chem. Int. Ed. 56 (2017) 4507-4511. DOI:10.1002/anie.201700196 |
| [11] |
E.R. Gillies, E. Dy, J.M.J. Fréchet, F.C. Szoka, Mol. Pharmaceutics 2 (2005) 129-138. DOI:10.1021/mp049886u |
| [12] |
L. Trachsel, M. Romio, B. Grob, et al., ACS Nano 14 (2020) 10054-10067. DOI:10.1021/acsnano.0c03239 |
| [13] |
W. Chen, Y. Zhou, Y. Li, et al., Polym. Chem. 7 (2016) 6789-6797. DOI:10.1039/C6PY01638C |
| [14] |
K. Zhang, G.N. Tew, React. Funct. Polym. 80 (2014) 40-47. DOI:10.1016/j.reactfunctpolym.2014.01.012 |
| [15] |
T. Josse, J. De Winter, P. Gerbaux, O. Coulembier, Angew. Chem. Int. Ed. 55 (2016) 13944-13958. DOI:10.1002/anie.201601677 |
| [16] |
H. Kammiyada, A. Konishi, M. Ouchi, M. Sawamoto, ACS Macro Lett. 2 (2013) 531-534. DOI:10.1021/mz400191t |
| [17] |
J. Huang, J. Li, R. Yan, et al., Chin. Chem. Lett. 35 (2024) 108643. DOI:10.1016/j.cclet.2023.108643 |
| [18] |
Y. Xia, A.J. Boydston, R.H. Grubbs, Angew. Chem. Int. Ed. 50 (2011) 5882-5885. DOI:10.1002/anie.201101860 |
| [19] |
A. Narumi, M. Yamada, Y. Unno, et al., ACS Macro Lett. 8 (2019) 634-638. DOI:10.1021/acsmacrolett.9b00308 |
| [20] |
S.A. Gonsales, T. Kubo, M.K. Flint, et al., J. Am. Chem. Soc. 138 (2016) 4996-4999. DOI:10.1021/jacs.6b00014 |
| [21] |
Y.A. Chang, R.M. Waymouth, J. Polym. Sci. A: Polym. Chem. 55 (2017) 2892-2902. DOI:10.1002/pola.28635 |
| [22] |
C.D. Roland, H. Li, K.A. Abboud, K.B. Wagener, A.S. Veige, Nat. Chem. 8 (2016) 791-796. DOI:10.1038/nchem.2516 |
| [23] |
Y.Y. Fei, C. Liu, G. Chen, C.Y. Hong, Polym. Chem. 10 (2019) 3895-3901. DOI:10.1039/c9py00472f |
| [24] |
C. Liu, Y.Y. Fei, H.L. Zhang, C.Y. Pan, C.Y. Hong, Macromolecules 52 (2019) 176-184. DOI:10.1021/acs.macromol.8b02192 |
| [25] |
H. Jacobson, W.H. Stockmayer, J. Chem. Phys. 18 (2004) 1600-1606. |
| [26] |
M. Ouchi, N. Badi, J.F. Lutz, M. Sawamoto, Nat. Chem. 3 (2011) 917-924. DOI:10.1038/nchem.1175 |
| [27] |
M. Gonzalez-Burgos, A. Latorre-Sanchez, J.A. Pomposo, Chem. Soc. Rev. 44 (2015) 6122-6142. DOI:10.1039/C5CS00209E |
| [28] |
S. Mavila, O. Eivgi, I. Berkovich, N.G. Lemcoff, Chem. Rev. 116 (2016) 878-961. DOI:10.1021/acs.chemrev.5b00290 |
| [29] |
A.M. Hanlon, C.K. Lyon, E.B. Berda, Macromolecules 49 (2016) 2-14. DOI:10.1021/acs.macromol.5b01456 |
| [30] |
Y. Hirai, T. Terashima, M. Takenaka, M. Sawamoto, Macromolecules 49 (2016) 5084-5091. DOI:10.1021/acs.macromol.6b01085 |
| [31] |
K. Matsumoto, T. Terashima, T. Sugita, M. Takenaka, M. Sawamoto, Macromolecules 49 (2016) 7917-7927. DOI:10.1021/acs.macromol.6b01702 |
| [32] |
H.L. Zhang, H. Zha, C. Liu, C.Y. Hong, Sci. China Chem. 65 (2022) 2558-2566. DOI:10.1007/s11426-022-1344-3 |
| [33] |
M. Shibata, M. Matsumoto, Y. Hirai, et al., Macromolecules 51 (2018) 3738-3745. DOI:10.1021/acs.macromol.8b00570 |
| [34] |
S. Imai, Y. Hirai, C. Nagao, M. Sawamoto, T. Terashima, Macromolecules 51 (2018) 398-409. DOI:10.1021/acs.macromol.7b01918 |
| [35] |
Y. Jiang, Z. Zhang, D. Wang, N. Hadjichristidis, Macromolecules 51 (2018) 3193-3202. DOI:10.1021/acs.macromol.8b00333 |
| [36] |
P. Sun, J.Q. Chen, J.A. Liu, K. Zhang, Macromolecules 50 (2017) 1463-1472. DOI:10.1021/acs.macromol.6b02614 |
| [37] |
X.Q. Xue, Y.J. Chen, K. Liang, et al., Polym. Chem. 11 (2020) 6529-6538. DOI:10.1039/d0py01116a |
| [38] |
B.A. Laurent, S.M. Grayson, J. Am. Chem. Soc. 133 (2011) 13421-13429. DOI:10.1021/ja2024355 |
| [39] |
V.V. Varadaiah, V.S.R. Rao, J. Polym. Sci. 50 (1961) 31-34. DOI:10.1002/pol.1961.1205015304 |
| [40] |
H.L. Zhang, W. Xu, C. Liu, C.Y. Hong, Polym. Chem. 12 (2021) 5357-5363. DOI:10.1039/d1py00341k |
| [41] |
M.L. McGraw, R.W. Clarke, E.Y.X. Chen, J. Am. Chem. Soc. 143 (2021) 3318-3322. DOI:10.1021/jacs.1c00561 |
| [42] |
Y. Muramatsu, A. Takasu, M. Higuchi, M. Hayashi, J. Polym. Sci. 58 (2020) 2936-2942. DOI:10.1002/pol.20200587 |
| [43] |
Z. Zhang, X. Nie, F. Wang, et al., Nat. Commun. 11 (2020) 3654. DOI:10.1038/s41467-020-17474-0 |
| [44] |
K. Zhang, M.A. Lackey, Y. Wu, G.N. Tew, J. Am. Chem. Soc. 133 (2011) 6906-6909. DOI:10.1021/ja2007559 |
| [45] |
C.H. Ma, Y. Quan, R.Y. Sun, et al., Chem. Commun. 58 (2022) 4340-4343. DOI:10.1039/d1cc07223d |
| [46] |
M. Schappacher, A. Deffieux, Angew. Chem. Int. Ed. 48 (2009) 5930-5933. DOI:10.1002/anie.200900704 |
| [47] |
M. Schappacher, A. Deffieux, Science 319 (2008) 1512-1515. DOI:10.1126/science.1153848 |