 2023, Vol. 34
2023, Vol. 34 


b School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China;
c Chinese Academy of Sciences, University of Chinese Academy of Sciences, Beijing 100049, China;
d Fujian College, University of Chinese Academy of Sciences, Fuzhou 350002, China
Ammonia (NH3) is an important industrial chemical used extensively in agricultural fuels, medicines, and fertilizers [1-3]. However, the traditional Haber-Basch process for ammonia synthesis has problems such as high energy consumption and serious pollution [4-6]. The electrochemical synthesis of ammonia has the characteristics of mild conditions, simple and safe process, high activity, and nitrogen reduction reaction (NRR: N2 + 6H+ + 6e− → 2NH3) can be carried out at ambient temperature and pressure [7-9]. Therefore, how to design and synthesize an efficient catalyst to reduce the activation energy of electrochemical NRR and inhibit the formation of hydrogen evolution reaction (HER: 2H+ + 2e− → H2) has become an important research focus[8]. Existing electro-catalytic ammonia synthesis catalysts include metal catalysts [7,8,10-18], non-metallic catalysts (such as boron-doped graphene, nitrogen-doped porous carbon, and single-boron) [19-21], polymers (such as polyaniline) [22], and composite catalysts (such as C18@Fe3P) [23], etc. Among them, metal catalysts can be divided into noble metal catalysts (such as Ru, Au, Pt, and their alloys) [8,10-12], and non-noble metal catalysts (such as Ni, Mo, and Fe) [7,13-16,24-27]. The high cost and rarity of traditional noble metallic catalysts limit their large application. Consequently, the development of high-activity non-noble metal catalysts has become a research hotspot. Zheng proved that heterogeneous Mo3 single-cluster catalysts supported on nitrogen-doped graphene can use for robust electrochemical nitrogen reduction [15]. Ma compared a series of transition metals TM3@BN (TM = V, Fe, Mo, W) [14], and found that Mo3 is an ideal NRR catalyst, which has the same catalytic ability as noble metal Au at the same time of low economic price.
The selection of catalyst carriers is equally important. From the perspective of different carriers' structural dimensions, common catalyst carriers include framework materials [28], two-dimensional lamellar materials (such as graphene, carbon nitrides, and transition metal carbides/nitrides) [25-27,29], etc. Hollow structural materials possess cavities in their structures, which can prolong the residence time of N2, enrich the reaction intermediates and shorten the electron transport path while stabilizing and exposing the active sites of the catalyst, thereby further enhancing its NRR activity [30]. Therefore, reasonable design of the pore size is very important. Pores with extremely small size are difficult for reactants to enter into the cavity [31], and are not conducive to anchoring catalytic active sites. Hollow structural materials with large specific area and high stability are suitable carriers. However, the hollow structure still has the problem of relying on the template method to synthesize, and it is difficult to obtain a precise and controllable structure. Crystalline aluminum-oxo clusters (AlOCs) possess precise molecular structure and excellent designability, which provide an ideal platform to study the catalytic effect. Zhang's group has reported a series of crystalline aluminum molecular rings with regular wheel-like structures [32], having the advantages of facile preparation [33], tunable sizes [34], and easy assembly [35]. Based on its size-adjustable cavities which can be used to nest metal clusters, aluminum molecular rings are an excellent catalyst carrier model.
Herein, we perform theoretical NRR calculations based on the largest aluminum-pyrazole ring Al10(OH)10(BA)6(PZ-CH3)14 (AlOC-69; BA = benzoate; PZ-CH3 = 4-methylpyrazole) to date. To simplify the calculation, the methyl groups on the pyrazoles are further replaced by H, constructing Al10O10. The d-orbitals of Mo and the p-orbitals of Al10O10 have formed stable Mo-O bonds, loading Mo3 into the center of Al10O10. DFT calculations show that Mo3@Al10O10 can activate N2 with a more suitable activation energy of the electrochemical NRR and can suppress the generation of the hydrogen evolution side reaction. The results show that Mo3@Al10O10 has catalytic performance comparable to that of noble metal catalysts and is a kind of good electrochemical NRR catalyst, providing a new approach for the design of efficient NRR catalysts with a novel structure.
To determine the stable structure of Mo3@Al10O10, several initial configurations were considered and the most stable configuration obtained after optimization is shown in Fig. 1a. The optimized structure of Mo3 is an approximately isosceles triangle, and two molybdenum atoms coordinate with two adjacent oxygen atoms in Al10O10 are approximately in parallel, forming two Mo-O bonds.

|
Download:
|
| Fig. 1. (a) Structure diagram of Mo3@Al10O10. (b) Charge density difference for Mo3@Al10O10. (c, d) Density of states diagram of Al10O10 and Mo3@Al10O10. | |
{kind=link}
The bond lengths of Mo1-Mo2, Mo1-Mo3, and Mo2-Mo3 are 2.375 Å, 2.296 Å, and 2.314 Å, respectively, and the bond lengths of Mo1-O1 and Mo2-O2 are 2.117 Å and 2.110 Å, respectively. The overall structure of Al10O10 has not undergone significant deformation. Thermodynamic stability is an important indicator of catalytic activity, and the strong interaction between catalyst and carrier can avoid the migration of metal atoms. The calculated formation free energy of Mo3@Al10O10 carrier is −0.76 eV. The result shows that there is a strong interaction between Mo3 and Al10O10, which shows Mo3 can be stabilized on the surface of the carriers.
To gain deeper insight into the stability of Mo3@Al10O10, its electronic structures were further analyzed. The charge density difference of Al10O10 before and after adsorbed Mo3 was calculated. As shown in Fig. 1b, the yellow and cyan lobes represent the accumulation and consumption of electrons in this area. It can be found that the Mo3 surface is mainly characterized by the reduction of electron density and the presence of positive electron charge, while the adjacent two oxygen surfaces have electron accumulation, which can prove that there is an obvious interaction between Mo-O. It proves that the Mo3 group can be stably adsorbed on Al10O10. Moreover, by observing the density of states (DOS) diagram before and after adsorption, as shown in Figs. 1c and d, both the d-orbitals of Mo3 and the p-orbitals of Al10O10, whether above or below the Fermi level, have a strong hybridization effect. It proves that there is an interaction between the d-orbitals of Mo3 and the p-orbitals of Al10O10. This electron transformation enables Mo3 to firmly coordinate with Al10O10 and form a stable catalytic structure.
The adsorption and activation of N2 on the catalyst is an important step in the whole NRR reaction process. The adsorption configuration and adsorption strength will directly affect the subsequent hydrogenation process. Through comparing the results of different structural optimization, the most stable configuration can be obtained (Fig. 2a). Through calculation, it is found that different from the end-on adsorption type of other metal catalysts, Mo3@Al10O10 adopts the side-on adsorption type at the upper of two Mo atoms. Compared with end-on adsorption (−0.90 eV), side-on adsorption has higher adsorption energy (−1.08 eV) and higher stability. At the same time, side-on adsorption can better activate N2 molecules. The bond lengths of Mo1-N1 and Mo3-N2 are 2.039 Å and 1.912 Å respectively. Nitrogen-nitrogen triple bond (N≡N) in N2 is weakened and extends from the original 1.105 Å to 1.261 Å, achieving the effect of activation.

|
Download:
|
| Fig. 2. (a) Structure diagram of adsorption of N2 on Mo3@Al10O10. (b) Charge density difference of the adsorption of N2 on Mo3@Al10O10. (c) Density of states diagram of Mo d-orbitals and N2 p-orbitals of N2 adsorbed Mo3@Al10O10. (d) Molecular orbital of N2-Mo3. | |
{kind=link}
The charge transfer mode can be analyzed from the charge density difference after N2 adsorption. As shown in Fig. 2b, N2 is the electron acceptor in general, but a small decrease in charge density can also be seen between N≡N, which indicates that the electron transfer between N2 and Mo3 is not unidirectional, and it will also feed back to Mo3 when N2 gets electrons. There are simultaneous two transfers of electrons, from the occupied σ-orbital electrons of N2 to the empty d-orbital of Mo, and from the occupied d-orbital of Mo to the π-antibonding-orbital of N2. The analysis of DOS (Fig. 2c) and molecular orbital of N2-Mo3 (Fig. 2d) also further proves that there is a strong interaction between N-Mo, and the p-orbitals of N overlap with the d-orbitals of Mo to form bonds. On this basis, the adsorbed N2 molecules are fully activated, and the first hydrogenation reaction is relatively easy to achieve.
By comparing different N2 reduction reaction modes, it is difficult to directly break N≡N under mild conditions. Consequently, NRR in this system is carried out in association mode. N2 is adsorbed on the surface of Mo3@Al10O10 to carry out the hydrogenation reaction before breaking N≡N to generate NH3. The Gibbs free energies of various paths without an applied electric field are calculated. As shown in Fig. 3, the free energy of adsorption of N2 adsorbed at Mo3@Al10O10 is −1.08 eV, indicating that this is a spontaneous thermodynamic process. The free energy of the process of forming *NH-N* intermediate is 0.19 eV, which is a non-spontaneous process. In the second hydrogenation process, 3A has lower free energy than 3B, which means process 3A is more energetically advantageous, and alternately H bonds on two N to generate *NH-NH* intermediate. *NH-NH* interacts with the third H to form *NH-NH2*. This process requires the highest energy, 0.28 eV, which is the limiting step of the total reaction. Then *NH-NH2* interacts with the fourth H and compared with process 5A adding on N2 to generate *NH2-NH2* (0.65 eV), the free energy of the process 5C adding on N1 to generate the first NH3 is negative (−1.34 eV), which is thermodynamic spontaneous, leaving the NH* intermediate. NH* then interacts with the fifth and sixth H to form NH2* intermediate and the second NH3*.

|
Download:
|
| Fig. 3. Reaction intermediates in the electroreduction of N2 to NH3 on Mo3@Al10O10 surface, with the corresponding ∆G (eV) in each step. | |
{kind=link}
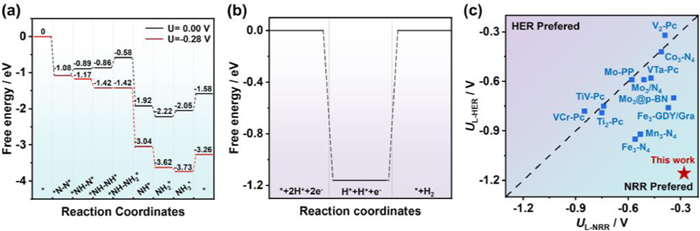
The limiting potential (UL) is given by −max(ΔG)/e, where ΔG is the free energy of each elemental protonation step in the NRR progress. As shown in Fig. 4a, only −0.28 V electric potentials are needed to make each step of the hydrogenation reaction become a thermodynamic spontaneous process, which is much lower than that of the most report (Fig. 4c and Table S6 in Supporting information) [7,14,18,29]. Since HER will consume H to generate H2, which is the main competitive reaction of NRR, the presence of HER will reduce the Faraday efficiency as well as the selectivity of NH3. Therefore, the free energy of HER on Mo3@Al10O10 was calculated as shown in Fig. 4b, it can be found that the energy barrier for the Heyrovsky step is 1.16 eV, which is far larger than the free energy of NRR (0.28 eV). Consequently, NH3 is the main product on Mo3@Al10O10, showing excellent selectivity.

|
Download:
|
| Fig. 4. (a) The calculated Gibbs free energy diagrams of the NRR on Mo3@Al10O10 at different applied potentials. (b) Gibbs free energy diagrams of the HER on Mo3@Al10O10. (c) Comparison of limiting potentials (UL) in this work and those of previous reports. | |
{kind=link}
In the present work, the largest aluminum-pyrazole ring (AlOC-69) to date was synthesized and combined with Mo3 to produce a promising NRR catalyst. The calculated formation energy (−0.76 eV) and the electron transfer between Mo-O show the strong interaction between Mo3 and Al10O10, demonstrating that Mo3 can be stabilized at the carrier surface. Meanwhile, Mo3@Al10O10 is an excellent NRR catalyst. The strong interaction between the p-orbital of N and the d-orbital of Mo makes the adsorbed N2 molecule fully activated, and the first hydrogenation reaction occurs easily. During the whole process, the protonation of *NH-NH* to *NH-NH2* is the limiting step with the maximum ΔG value of 0.28 eV, which is much smaller than competing HER (1.16 eV), showed promising catalytic selectivity. The precise molecular structure provides the basis for studying its structure-activity relationship and this novel structure breaks through the limitations of catalyst carriers, opening a new path to advance the sustainable production of NH3.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 92161105 and 92061104), Natural Science Foundation of Fujian Province (Nos. 2021J06035 and 2021J01525) and Youth Innovation Promotion Association CAS (No. Y2021081).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108604.
| [1] |
T. Spatzal, K.A. Perez, O. Einsle, J.B. Howard, D.C. Rees, Science 345 (2014) 1620-1623. DOI:10.1126/science.1256679 |
| [2] |
R.F. Service, Science 345 (2014) 610-610. DOI:10.1126/science.345.6197.610 |
| [3] |
K.A. Brown, D.F. Harris, M.B. Wilker, et al., Science 352 (2016) 448-450. DOI:10.1126/science.aaf2091 |
| [4] |
F. Bozso, G. Ertl, M. Weiss, J. Catal. 50 (1977) 519-529. DOI:10.1016/0021-9517(77)90063-X |
| [5] |
M. Kitano, Y. Inoue, Y. Yamazaki, et al., Nat. Chem. 4 (2012) 934-940. DOI:10.1038/nchem.1476 |
| [6] |
C.J.M. van der Ham, M.T.M. Koper, D.G.H. Hetterscheid, Chem. Soc. Rev. 43 (2014) 5183-5191. DOI:10.1039/C4CS00085D |
| [7] |
L. Zhang, X. Ji, X. Ren, et al., Adv. Mater. 30 (2018) e1800191. DOI:10.1002/adma.201800191 |
| [8] |
C. Ling, Y. Zhang, Q. Li, et al., J. Am. Chem. Soc. 141 (2019) 18264-18270. DOI:10.1021/jacs.9b09232 |
| [9] |
Y. Wang, N. Yang, X. Xin, et al., Chin. Chem. Lett. 34 (2023) 107841. DOI:10.1016/j.cclet.2022.107841 |
| [10] |
M.M. Shi, D. Bao, B.R. Wulan, et al., Adv. Mater. 29 (2017) 1606550. DOI:10.1002/adma.201606550 |
| [11] |
M. Nazemi, S.R. Panikkanvalappil, M.A. El-Sayed, Nano Energy 49 (2018) 316-323. DOI:10.1016/j.nanoen.2018.04.039 |
| [12] |
V. Kordali, G. Kyriacou, C. Lambrou, Chem. Commun. (2000) 1673-1674. |
| [13] |
S. Licht, B. Cui, B. Wang, et al., Science 345 (2014) 637-640. DOI:10.1126/science.1254234 |
| [14] |
S. Gao, Z. Ma, C. Xiao, et al., J. Mater. Sci. Technol. 108 (2022) 46-53. DOI:10.1016/j.jmst.2021.08.052 |
| [15] |
G. Zheng, L. Li, Z. Tian, X. Zhang, L. Chen, J. Energy Chem. 54 (2021) 612-619. DOI:10.1016/j.jechem.2020.06.048 |
| [16] |
C. Cui, H. Zhang, R. Cheng, B. Huang, Z. Luo, ACS Catal. 12 (2022) 14964-14975. DOI:10.1021/acscatal.2c04146 |
| [17] |
X.L. Ma, M. Li, J.B. Lu, C.Q. Xu, J. Li, Chin. J. Struct. Chem. 41 (2022) 2212080-2212088. |
| [18] |
X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec, S.Z. Qiao, J. Am. Chem. Soc. 141 (2019) 9664-9672. DOI:10.1021/jacs.9b03811 |
| [19] |
X. Yu, P. Han, Z. Wei, et al., Joule 2 (2018) 1610-1622. DOI:10.1016/j.joule.2018.06.007 |
| [20] |
X. Yang, K. Li, D. Cheng, et al., J. Mater. Chem. A 6 (2018) 7762-7769. DOI:10.1039/C8TA01078A |
| [21] |
C. Zhao, S. Zhang, M. Han, et al., ACS Energy Lett. 4 (2019) 377-383. DOI:10.1021/acsenergylett.8b02138 |
| [22] |
J. Yu, J. Li, X. Zhu, et al., ChemElectroChem 6 (2019) 2215-2218. DOI:10.1002/celc.201900320 |
| [23] |
T. Xu, J. Liang, Y. Wang, et al., Nano Res. 15 (2022) 1039-1046. DOI:10.1007/s12274-021-3592-8 |
| [24] |
Y.B. Li, Y.P. Liu, J. Wang, Y.L. Guo, K. Chu, Inorg. Chem. Front. 7 (2020) 455-463. DOI:10.1039/C9QI01133A |
| [25] |
J. Wang, M. Shi, G. Yi, et al., Chin. Chem. Lett. 33 (2022) 4623-4627. DOI:10.1016/j.cclet.2021.12.040 |
| [26] |
H.Q. Yin, L.L. Yang, H. Sun, et al., Chin. Chem. Lett. 34 (2023) 107337. DOI:10.1016/j.cclet.2022.03.060 |
| [27] |
S. Wu, M. Zhang, S. Huang, et al., Chin. Chem. Lett. 34 (2023) 107282. DOI:10.1016/j.cclet.2022.03.005 |
| [28] |
H.K. Lee, C.S.L. Koh, Y.H. Lee, et al., Sci. Adv. 4 (2018) eaar3208. DOI:10.1126/sciadv.aar3208 |
| [29] |
X. Wang, Q. Zhang, W. Hao, et al., J. Mater. Chem. A 10 (2022) 15036-15050. DOI:10.1039/D2TA02887E |
| [30] |
C. Xue, X. Zhou, X. Li, et al., Adv. Sci. 9 (2022) 2104183. DOI:10.1002/advs.202104183 |
| [31] |
M. Nazemi, M.A. El-Sayed, J. Phys. Chem. Lett. 9 (2018) 5160-5166. DOI:10.1021/acs.jpclett.8b02188 |
| [32] |
Y. Li, C. Zheng, S.T. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202116563. DOI:10.1002/anie.202116563 |
| [33] |
S.T. Wang, Y.J. Liu, C.C. Feng, W.H. Fang, J. Zhang, Aggregate 4 (2023) e264. DOI:10.1002/agt2.264 |
| [34] |
L. Geng, C.H. Liu, S.T. Wang, W.H. Fang, J. Zhang, Angew. Chem. Int. Ed. 59 (2020) 16735-16740. DOI:10.1002/anie.202007270 |
| [35] |
S. Yao, W.H. Fang, Y. Sun, S.T. Wang, J. Zhang, J. Am. Chem. Soc. 143 (2021) 2325-2330. DOI:10.1021/jacs.0c11778 |