 2023, Vol. 34
2023, Vol. 34 


b School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China;
c NHC Key Laboratory of Nuclear Medicine, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China;
d Department of Radiation Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450000, China;
e School of Materials and the Environment, Beijing Institute of Technology, Zhuhai 519085, China;
f Rigerna Therapeutics, Suzhou 215127, China
Small interfering RNAs (siRNAs) can silence almost any target gene in complete base pairs complementary manner [1], such process can effectively cure diseases due to proteins’ abnormal high expression [2] or malfunction. Thus, RNAi therapy is considered one of the most promising biopharmaceutical technologies after small molecule drugs and monoclonal antibodies, because it offers a treatment strategy for diseases that seem undruggable and untargetable. Unfortunately, it still faces many challenges, especially the lack of ideal carriers [3]. To address this, numerous vehicles were developed, including lipid or lipidoid nanoparticles (LNPs) [4-6], polymers [7,8] or dendrimers [9,10], exosomes [11], peptides [12-15], DNA carriers [16-18], inorganic carriers [19] or others. Among them, LNPs are a significant class of siRNA delivery systems because they show high siRNA encapsulation capacity and satisfactory delivery efficiency. OnpattroⓇ (Alnylam), the first commercially approved siRNA therapeutic, demonstrated the high gene silence potency and prolonged blood circulation that LNPs can achieve. But the high level of unnatural nanomaterials is considered the leading cause of toxicity [20] and reduces the ratio between drug and carrier. Besides, weak electrostatic interactions between the vehicle and siRNA seemed readily cause “burst release” after systemic administration [21].
The new clinical programs all used another class of strategy instead of LNPs, it employs a linker conjugating the delicate modified siRNAs to the functional group directly. This strategy significantly increased the ratio between drug and carrier. More importantly, these functional groups naturally exist in living organisms, thus, they can avoid adverse effects to a great extent. After the combination with siRNAs, the functional groups significantly improved the performance of siRNA therapeutics by exerting their biological functions. Among those strategies, the N-acetylgalactosamine (GalNAc) conjugated siRNAs leverage high affinity between GalNAc and its receptor (asialoglycoprotein receptor, ASGPR), almost offers a perfect delivery scheme for delivering siRNA to hepatocytes. ASGPRs are highly and specifically expressed in the basolateral membrane of hepatocytes [22], and they can efficiently transport galactose-derived ligands, including GalNAc, from the cell surface to the cytoplasm of hepatocytes [23]. LeqvioⓇ (Novartis) is a commercialized GalNAc conjugated siRNA therapeutic that targets proprotein convertase subtilisin kexin type 9 (PCSK9), it showed astonishing performance in a clinical test. The results showed that LeqvioⓇ supports the dosing regimen every 6 months after two doses. While ensuring effectiveness, it is expected to solve the long-term compliance dilemma of patients. Since Alnylam announced their GalNAc conjugated delivery program, most siRNA pharmaceutical companies have developed their delivery platforms based on GalNAc for delivering siRNA to hepatocytes. More recently, Alnylam conjugated 2′-O-hexadecyl at position 6 from 5′ of the sense strand of siRNA, enabling potent and durable gene silence in the central nervous system (CNS), eye and lung in animals [24]. Indeed, this scheme expanded RNAi therapeutics to extrahepatic tissues, however, efficiently delivering siRNA to tumor is still challenging.
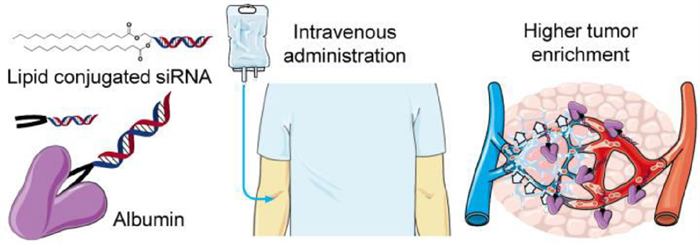
Albumin is a major soluble protein throughout blood circulation and has a notable circulation halftime (T1/2) of around twenty days [25]. Albumin is synthesized in liver and secreted into circulation. It is regarded as an ideal carrier [26] because it readily covalent attaches to, non-covalent associates with, or encapsulates [27] poorly water-soluble molecules [28], rescuing drugs from systemic clearance and degradation. In addition, since albumin can accumulate in tumor tissues by enhanced permeability and retention (EPR) effect and its receptor glycoprotein 60 (gp60) [27], it was already considered for cancer treatment. However, the investigations about albumin-based carriers were only around small molecules and protein therapeutics, studies about lipophilic siRNA associate with albumin are still limited. Herein, we synthesized lipid conjugated siRNAs (LsiRNAs), which offer two octadecyl chains that can insert into the hydrophobic core of albumin by non-covalent association, leading to high-affinity to albumin (Fig. 1). After this interaction, LsiRNAs extremely prolonged its half-life in vivo and increased tumor retention. Compared with nanoparticle-encapsulated siRNA, LsiRNA did not significantly reduce the efficiency of gene silence but resulted in much better safety. Importantly, LsiRNA against programmed death 1 ligand 1 (LsiPdl1) showed prominent antitumor ability on tumor-bearing mice. It was observed that 10 mg/kg LsiPdl1 effectively increased infiltration of CD8+ T cells and promoted the maturation of dendritic cells. In brief, our study provides a simple but effective way for siRNA tumor delivery that “hitchhikes” on long-circulating albumin.

|
Download:
|
| Fig. 1. Schematic representation of the study. The lipid conjugated siRNA can efficiently non-covalent bind to the hydrophobic core of albumin, thereby increasing the siRNA enrichment within the tumor. | |
{kind=link}
The LsiRNAs target hepatitis B virus (HBV, LsiHBV) and programmed death 1 ligand 1 (Pdl1, LsiPdl1) are used in this study. The synthesis procedures are described in Fig. 2A and the section “Materials and methods” in Supporting information, the confirmation are shown in supplementary materials (Figs. S1 and S2 in Supporting information).

|
Download:
|
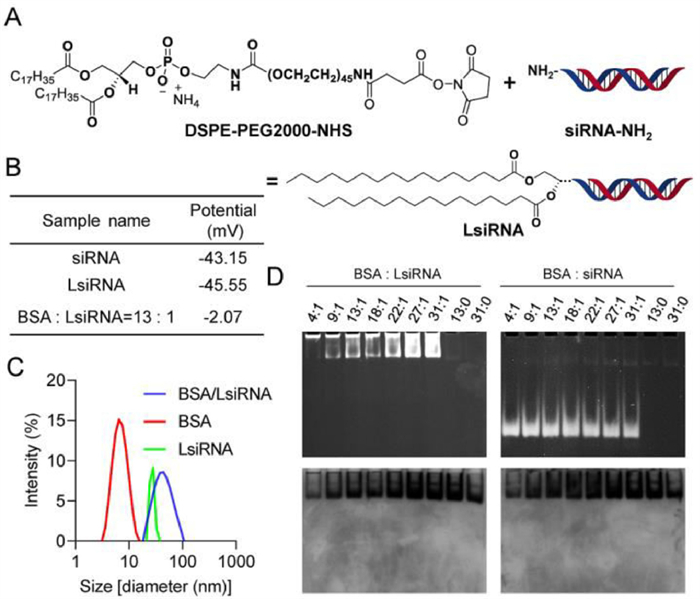
| Fig. 2. LsiRNA synthesis strategy and its binding test with BSA. (A) The synthesis strategy of LsiRNA. (B) Zeta potential and (C) particle size of LsiRNA before and after the incubation with BSA, measured by DLS. (D) Gel retardation was used to determine the interaction between BSA and LsiRNA, imaging for siRNA or LsiRNA (top) and BSA (bottom). | |
{kind=link}
Ensuring stability is a key premise for siRNA to function in vivo. Thus, we tested the stability of LsiHBV and LsiPdl1 after the synthesis. Here, LsiRNAs were incubated with human serum, mouse serum and fetal bovine serum (FBS) at a volume ratio of 1:9 and 37 ℃, respectively. After sampling at different times, the stability of LsiRNAs was tested by polyacrylamide gel electrophoresis (PAGE). The results showed that all two LsiRNAs maintained stable in human serum, mouse serum and fetal bovine serum (Fig. S3 in Supporting information). These results supported further application of LsiRNA in vivo.
To test whether LsiRNAs binds to albumin, several studies were carried out. Firstly, the zeta potential and the size of free LsiRNA and its complex with bovine serum albumin (BSA) were determined by dynamic light scattering (DLS). Data showed that lipid conjugated siRNA had strong electronegativity (−43.15 mV), similar to unconjugated siRNA. However, the zeta potential of the complex increased with the increase in BSA, (data not shown). Until the ratio of BSA:LsiRNA reached 13:1, the complex showed uncharged (Fig. 2B). Considering BSA also showed electronegativity at neutral pH, we assume that this potential change is caused by the shielding of the negatively charged group after the comination of both. Meanwhile, the size variation also indicated the successful binding of LsiRNA with BSA (Fig. 2C). The data showed that the size of free LsiRNA micelle and BSA were around 28 nm and 6 nm, respectively, and the complex of both was around 70 nm. Besides, we also performed a gel retardation assay on the complex as mentioned above to further test albumin-binding capacity. The agarose gel was first imaged for nucleic acid at 320 nm, then stained with Coomassie blue to show the migration of albumin. The result (Fig. 2D) showed that LsiRNA and albumin migrated in the exact location, proving the binding of BSA and LsiRNA. In contrast, BSA and siRNA without lipid conjugate were located in a different location. By far, the binding between LsiRNA and albumin was confirmed in several ways.
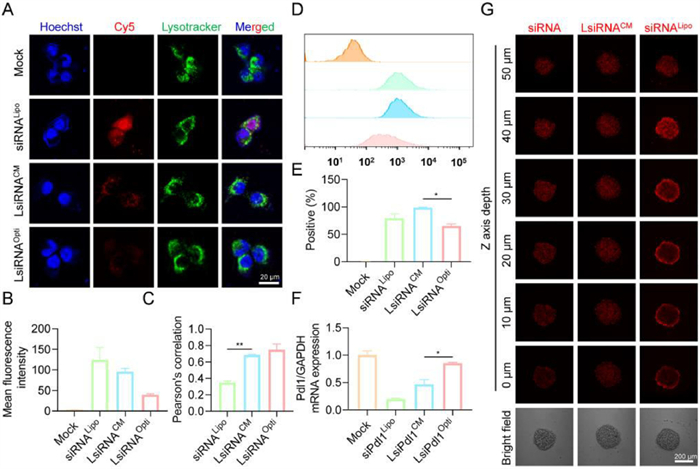
After confirming that LsiRNA binds to albumin, we sought to understand whether albumin is necessary for the cellular uptake of LsiRNA. To determine that, 4T1-Luc cells were seeded into dishes for further treatment. Cy5 labeled LsiHBV (Cy5-LsiHBV) was tested on cells cultured in albumin-presenting medium (complete medium) and albumin-free medium (Opti-MEM), Lipofectamine 2000 (Lipo 2000) formulated Cy5-siHBV were treated to cells cultured in Opti-MEM as positive control. After four hours of transfection, the treated cells were collected for confocal microscopy observation. It can be seen that Cy5-LsiHBV were efficiently endocytosed by cells in albumin-presenting environment (LsiRNACM), but the efficiency is lower than that mediated by Lipo 2000 (siRNALipo) (Figs. 3A and B). However, compared to that, cells treated with LsiHBV but culture in albumin-free medium (LsiRNAOpti) showed barely fluorescence signals (Figs. 3A and B), suggesting that albumin is essential for LsiRNA cellular uptake. In addition, we also analyzed the co-efficiency between LsiRNA and endosome or lysosome. Though LsiHBV do not contain any modules to facilitate endosome or lysosome escape, the data proved that part LsiHBV was successfully leaked from endosome or lysosome (Fig. 3C), we infer that is mainly due to the structural destruction of endosomal or lysosomal membrane. However, it also should be noted that the Pearson’s correlation of LsiHBV is much higher than Lipo 2000, which means more LsiRNA cannot function in cytoplasm. To further explore the endocytosis efficiency and the manner of LsiRNA, the treated cells were also determined by using fluorescence-activated cell sorting (FACS). The results further confirmed that LsiRNACM were efficiently endocytosed by cells, it can be seen that the endocytosis efficiency of LsiRNA is comparable to that of Lipo 2000 (Fig. 3D), and the positive endocytosis rate is higher than siRNALipo (Fig. 3E). Subsequently, the delivery efficiency was quantified by gene expression determination (Fig. 3F). Herein, siPdl1 and LsiPdl1 were used. The result showed that siPdl1 delivered by Lipo 2000 achieved 80.2% downregulation of Pdl1 expression at siRNA concentration of 200 nmol/L, LsiPdl1 silenced 53.5% of Pdl1 expression at the same siRNA concentration, whereas LsiRNA in Opti-MEM almost impossible to suppress mRNA expression. In addition, cell viabilities were evaluated, LsiHBV showed excellent safety in vitro (Fig. S4 in Supporting information). Notably, the viability of 3000 nmol/L LsiHBV-treated cells was satisfactory, which was 85.6% compared with phosphate buffer solution (PBS)-treated cells.

|
Download:
|
| Fig. 3. The internalization and penetration investigation of LsiRNA. (A) Subcellular localization of LsiRNA, cells were cultured in albumin-presenting medium (LsiRNACM) and albumin-free medium (LsiRNAOpti), siRNA transfected by Lipo 2000 (siRNALipo) was set as positive control. (B) Mean fluorescence intensities recorded in (A). (C) Co-efficiencies between LsiRNA or siRNA and endosome or lysosome. (D) Cellular uptake efficiency and (E) endocytosis positive rate, analyzed by FACS. (F) Gene expression determination, concentration of siRNA or LsiRNA was 200 nm. (G) Penetration comparation between LsiRNACM and siRNALipo, recorded by confocal microscope. Each bar represents the mean ± standard error of mean (S.E.M.), n = 2, *P < 0.05, **P < 0.01. | |
{kind=link}
The relatively large size of nanoparticles (>100 nm) hampers them from sufficient penetration into tumors and limits the treatment outcomes of nanomedicines [29]. Thus, the smaller albumin/LsiRNA is expected to increase tumor permeability compared to nanoparticles and may lead to better anti-tumor performance. Here, the permeability of albumin/LsiRNA was evaluated by 3D multicellular tumor spheroids (MCS) in vitro (Fig. 3G). After treating cells with Cy5-LsiHBV or Cy5-siHBV@Lipo 2000 for four hours, the permeability of two test substances was evaluated by confocal microscopy. It can be seen that the signals of Cy5-LsiHBV were homogeneously distributed throughout the tumor spheroid. In contrast, though the fluorescent signals of Cy5-siHBV@Lipo 2000 seem stronger than that of Cy5-LsiHBV, most Lipo 2000 nanoparticles were trapped at the surface of tumor spheroid, and only a few fluorescent signals can be detected at the core of the tumor spheroid. Besides, the penetration of free Cy5-siHBV was also tested. It can be seen that Cy5-siHBV also showed homogeneous penetration. However, considering that free siRNAs are hard to endocytose by cells, these free siRNAs only gathered outside the cells and would not function at cytoplasm. Such results proved that albumin/LsiRNA has better penetration than common nanoparticles.
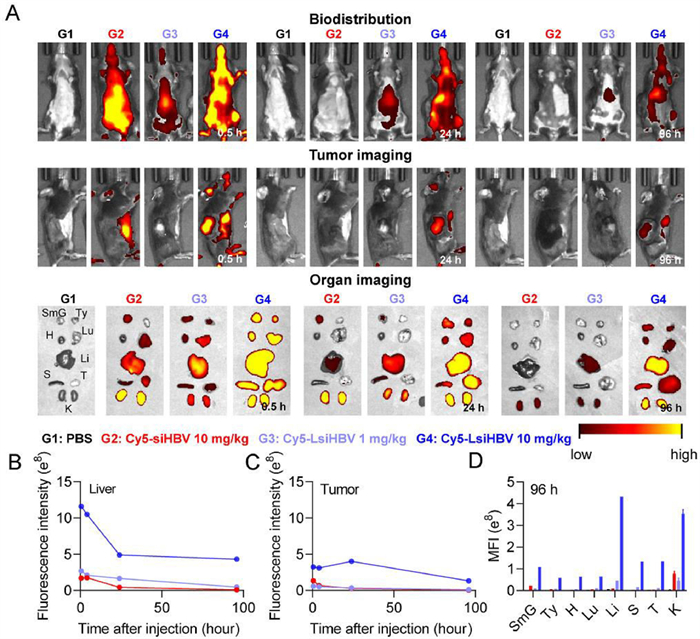
After in vitro analysis, we try to understand the biodistribution manner of LsiRNA. In this work, animal welfare and all experimental protocols have been reviewed and approved by the Animal Ethics Committee of Beijing Institute of Technology. Here, 16 Hepa1–6 xenograft tumor-bearing C57BL/6 mice were used as animal model and divided into 4 groups as follows: 1 × PBS, Cy5-siHBV 10 mg/kg, Cy5-LsiHBV 1 mg/kg and Cy5-LsiHBV 10 mg/kg. After single intravenous administration, animals were subjected to living imaging at 0.5, 2, 4, 6, 8, 24, 48, 72 and 96 h post-administration (Fig. 4A and Fig. S5 in Supporting information). In addition, after the living imaging at 0.5, 4, 24 and 96 h after administration, one animal from each group was sacrificed for organ imaging (Fig. 4A and Fig. S6 in Supporting information). The image data proved that Cy5-LsiHBV showed dose-dependent manner of fluorescent signals. Additionally, it can be seen that Cy5-LsiHBV quickly diffused throughout the body during the first 0.5 h, whereas Cy5-siHBV is mainly enriched in kidneys. Together with all living images, it is easy to figure out that albumin extremely increased the enrichment of LsiHBV in liver and tumor (Figs. 4B and C, and Fig. S5 in Supporting information). Not only the enrichment in liver and tumor of Cy5-LsiHBV 10 mg/kg was significantly higher than that of Cy5-siHBV 10 mg/kg, even the signal of Cy5-LsiHBV 1 mg/kg was also higher than that of Cy5-siHBV 10 mg/kg during the most of observation period. Besides, albumin also significantly prolonged LsiHBV retention in tumors, where Cy5-LsiHBV existed in tumors at least for 96 h but Cy5-siHBV only existed in tumors about 24 h (Figs. 4A and C, and Fig. S7 in Supporting information). Delightfully, the average fluorescence intensity in the tumor continuously increased during the first 24 h after administration, which was almost the same as that in liver at 24 h time point (Fig. 4C). After that, the average fluorescence intensity in the tumor began to decrease, but remained at a higher level than in most of the other organs. In addition, we also quantified of fluorescence signals in order to analyze their proportions in different organs. General view, the proportion of Cy5-LsiHBV signals in tumor showed an increasing trend (Fig. 4D and Fig. S8 in Supporting information), which increases from 3.2% at 0.5 h to 6.2% at 96 h in animals received Cy5-LsiHBV 1 mg/kg, besides, the percentage in tumor of Cy5-LsiHBV 10 mg/kg was 6.6% at 0.5 h, 6.9% at 4 h. It peaked at 16.8% at 24 h and remained at 16.0% at 96 h. In contrast, the proportion of Cy5-siRNA signals in tumor has remained steady at around 3% throughout the test. Collectively, due to the high affinity to albumin, LsiRNA showed significantly longer duration and higher retention than free siRNA, especially in tumor and liver. In addition, LsiRNA caused no toxicity up to 10 mg/kg in vivo, which no adverse effects were observed during the test.

|
Download:
|
| Fig. 4. Biodistribution test of LsiRNA on Hepa1–6 xenograft tumor-bearing C57BL/6 mice. (A) The living images and organ images were collected at 0.5, 24 and 96 h post-administration. SmG, submandibular gland; Ty, thymus; H, heart; Lu, lung; Li, liver; S, spleen; T, tumor; K, kidney. Average fluorescence intensity in (B) liver and (C) tumor at different time was recorded by IVIS Spectrum CT (PerkinElmer, US) and analyzed by software called LivingImage. (D) Quantitative analysis of the fluorescence signals in isolated organs at 96 h post-dose. | |
{kind=link}
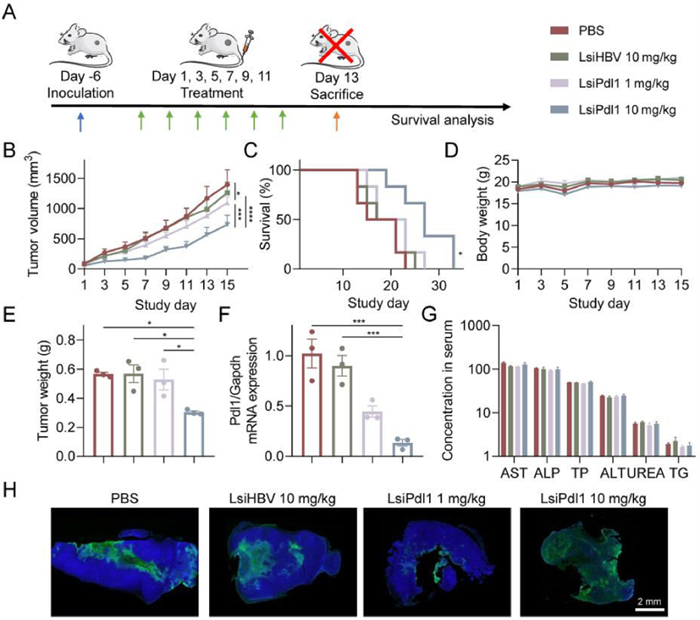
After confirming that LsiRNAs have excellent tumor retention capacity, we sought to investigate its therapeutic efficacy by using LsiPdl1 as therapeutic unit, since high expressed Pdl1 cause immune checkpoint escape of tumor cells. Besides, Pdl1 gene is regarded as an ideal therapeutic target, because it rarely expresses in most normal tissues but highly expresses on the surface of most tumor cells [30]. Herein, BALB/c mice were subcutaneously inoculated with 4T1-Luc cells, then randomly divided into four groups after six days as follows: (1) PBS, (2) LsiHBV 10 mg/kg, (3) LsiPdl1 1 mg/kg and (4) LsiPdl1 10 mg/kg. The formulations were intravenously injected every other day for six doses (Fig. 5A). The tumor volumes, survival rates and body weights were monitored throughout the study to evaluate the antitumor outcomes (Figs. 5B–D, Fig. S9 in Supporting information). It can be seen that 10 mg/kg LsiPdl1 suppressed tumor growth, extended the survival period significantly, and showed no toxicity which was reflected by body weight changes. Besides, 1 mg/kg LsiPdl1 slightly reduced tumor growth and prolonged treated animals’ life span. LsiHBV leads to no treatment outcomes compared with PBS, suggesting that all treatment outcomes were mediated by Pdl1 but not the toxicity of the formulation. In addition to what was mentioned above, three mice from each group were euthanized 48 h after the last treatment to further investigate the performance of formulations. In line with previous data, the data of tumor weights (Fig. 5E), real-time quantitative polymerase chain reaction (qPCR, Fig. 5F), serum biochemical analysis (Fig. 5G and Fig. S10 in Supporting information) and other indicators (Figs. S11 and S12 in Supporting information) also showed that LsiPdl1 resulted in tumor suppression by inhibiting target gene and no toxicity. Notably, due to the excellent tumor penetration and retention of LsiRNA, LsiPdl1 significantly mediated tumor apoptosis (Fig. 5H and Fig. S13 in Supporting information). The terminal dexynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay and hematoxylin and eosin (H&E) staining visually showed that 10 mg/kg LsiPdl1 induced structural changes and apoptosis in almost whole tumor. In contrast, apoptosis can only be detected in a few parts of tumors treated by PBS or LsiHBV or 1 mg/kg LsiPdl1.

|
Download:
|
| Fig. 5. Tumor suppression test of LsiPdl1. (A) Schematic illustration of the treating schedule. (B) Tumor growth curves, (C) survival curves and (D) body weight changes of all treated groups, n = 6. (E) Tumor weight, (F) relative Pdl1 mRNA expression and (G) main serum biochemical indicators of treated animals, n = 3. The animals were euthanatized at two days after the last dose. Six parameters including aspartate aminotransferase (AST, U/liter), alkaline phosphatase (ALP, U/liter), total protein (TP, g/liter), alanine aminotransferase (ALT, U/liter), urea nitrogen (UREA, mol/L), and TG (mmol/L) were recorded. (H) TUNEL staining of tumor sections. Each bar represents the mean ± S.E.M. *P < 0.05, ***P < 0.001, ****P < 0.0001. | |
{kind=link}
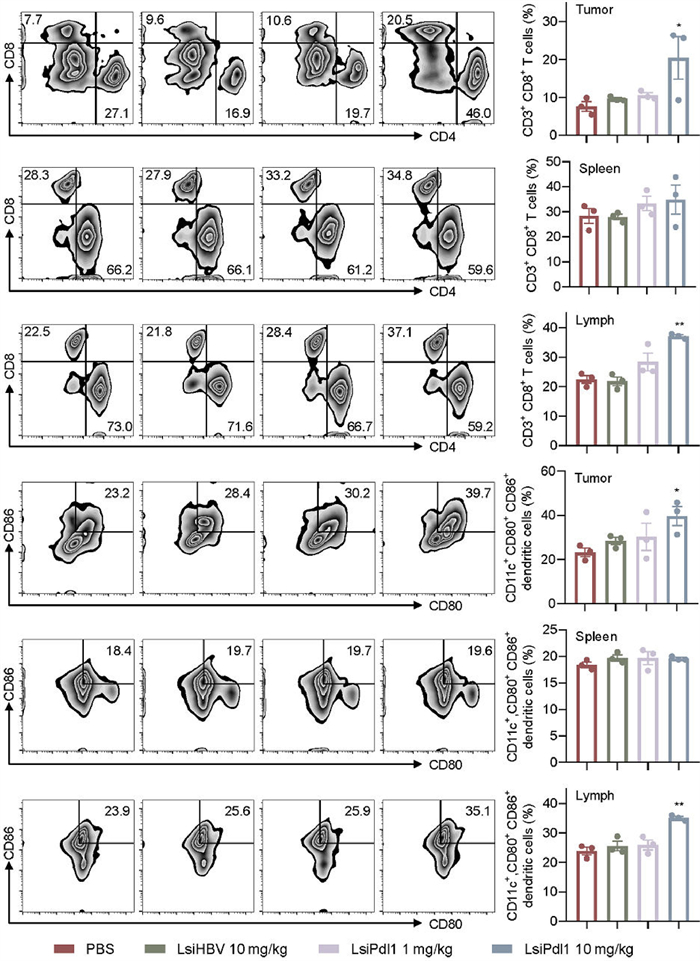
According to the references, Pdl1 not only regulates the infiltration of CD8+ T cells in tumor tissues, also binds to CD80 to inhibit dendritic cells (DCs) maturation and antigen presentation [31,32]. To test whether LsiPdl1 function by increasing the infiltration of CD8+ T cells and promoting DCs maturation, three animals of each group were euthanized after corresponding treatments. To fully explore the function manner of LsiPdl1, tumor cells, spleen cells and lymphocytes were collected and the level of CD4+ T cells, CD8+ T cells and CD80+ CD86+ DCs were determined by FACS (Fig. 6). The results showed that 10 mg/kg LsiPdl1 significantly increased the infiltration of CD4+ T cells and CD8+ T cells as well as promoted DCs maturation in tumor. Similarly, the level of CD8+ T cells significantly increased in the lymph nodes in 10 mg/kg LsiPdl1 treated animals. However, the drug did not affect T cells and DCs in the spleen. In addition, animals treated with 1 mg/kg LsiPdl1 produced no significant immune response in vivo, and LsiHBV resulted no immune response. Taken together, high level of LsiPdl1 efficiently suppressed tumor growth by increasing CD8+ T cells and promoting DCs maturation in tumor and lymph.

|
Download:
|
| Fig. 6. Immune response in tumor, spleen and lymph of treat animals. Each bar represents the mean ± S.E.M. *P < 0.05, **P < 0.01 vs. PBS group. | |
{kind=link}
Simply conjugating lipid to siRNA greatly improved the association between siRNAs and albumin, resulting in longer blood circulation, lower renal clearance, greater tumor retention and penetration, and higher cellular internalization of siRNAs.
The siRNAs hold greater potential than traditional drugs due to their function manner. The way that siRNAs interact with mRNAs, which are located more upstream than proteins in central dogma, endows siRNAs to be more targetable and druggable [33]. However, defect like unsatisfied pharmacokinetic characters hinder the usage of siRNAs. Due to instability in circulation and small size, siRNAs are often completely encapsulated into nanoparticles to protect them from nuclease attack and renal clearance. The nanoparticles not only mediated efficient liver siRNA delivery but also led to unintended toxicity due to their exogenous and complicated materials [34]. Many efforts were proposed to keep siRNA stable in blood, modification strategies represented by standard template chemistry (STC), enhanced stabilization chemistry (ESC) and others continually increased the RNases resistance of siRNAs. These improvements made shells (nanoparticles) are seemed no longer necessary and gave rise to another great revolution. By combining chemical modification strategy and GalNAc conjugation technology, formulations showed unmatched pharmacokinetic properties, safety and extremely high gene silencing efficiency. This approach almost perfectly solved the problem of siRNA liver delivery. Besides, the introduction of 2′-O-hexadecyl in siRNA resulted in robust and durable gene silence in CNS, eye and lungs. However, if the view enlarged to major organs and tissues, including tumor, the delivery problem is far from solved. Currently, tumor siRNA therapeutics in clinic mostly delivered by LNPs or polymers, these vehicles are mainly based upon EPR effect to increase drug tumor retention. However, the contribution of the EPR effect to drug enrichment in tumor is limited, report have shown that only 0.7% of the siRNA were successfully delivered to tumor tissue [35]. The presence of EPR effect in human tumors is controversial because the permeable nature of commonly used mouse tumor models is quite different from human tumors [36,37]. Some strategies sought to add target motifs on the surface of carrier but also led to minimal benefits. These facts explained why the development of siRNA tumor therapy is not so desirable.
As similar with GalNAc, albumin can be used as an endogenous carrier. Besides, it also featured as long-circulating and easy to accumulate in tumor. The example of nab-paclitaxel [38] proved that albumin could be used for tumor delivery in the clinic, and lead to higher tolerance dose than paclitaxel [39]. Herein, following the concept of GalNAc–siRNA conjugates, we conjugated modified siRNA to two alkyl tails. By association with albumin in vivo, LsiRNA avoided renal clearance and showed the tendency to tumors, which paves the way of tumor therapeutic. The biodistribution test showed that the intensity in the body and circulation half-life of LsiRNA was significantly higher than free siRNA. More importantly, it was also observed that LsiRNA high leveled and long period accumulated in tumor. We believe these data support the promise of LsiRNA as a drug for development.
Safety is another challenge for siRNA delivery. As mentioned above, nanoparticles are usually assembled by complicated components, easily leading to acute toxicity and irreversible liver damage. However, LsiRNAs consist only of nucleotides and DSPE-PEG2000, both of which are biocompatible. As expected, LsiRNAs exhibited excellent safety both in vitro and in vivo. It was shown that the concentration of 3000 nmol/L in vitro and 10 mg/kg in vivo caused no toxic side effects. It is anticipated to have very broad therapeutic windows according to these data.
The effectiveness of anticancer siRNA therapeutics is also affected by penetration depth into the tumor tissue. Owing to low diffusion efficiency and high interstitial pressure inside solid tumors, common nanocarrier usually showed poor penetration capacity and only trapped around tumor surface [40-42]. In contrast, in observations among a variety of tumor models, permeability to albumin is around 4-fold greater than 100 nm liposomes [43]. In line with the above statement, LsiRNAs penetrated deeper than Lipo 2000 (representing nanoparticles), the former achieved homogeneous distribution throughout the tumor spheroids but the latter failed to penetrate the core of tumor tissues. Researchers have claimed that intravenously administered nanomedicine must efficiently go through five cascades to achieve efficient tumor treatment: circulation in the blood, accumulation in tumor, penetration deep into the tissue, internalization into cells and release to the cytoplasm [44]. Compared with nanoparticles, LsiRNAs have clear advantages in the first three steps, especially in the second and the third steps. However, there is no motif in LsiRNAs promotes endocytosis and endosomal escape, thus, they have some disadvantages in the latter two steps. In vitro qPCR tests demonstrated that LsiRNAs were about 70% as efficient as Lipo 2000. However, in the complex environment in the body, LsiRNAs with orders of magnitude advantage in tumor not only can compensate for this disadvantage but also achieve more efficient target gene inhibition in tumor. In tumor suppression test, 10 mg/kg LsiPdl1 mediated about 87% target gene inhibition, and 1 mg/kg LsiPdl1 reduced relative mRNA expression by about 56% after doses. This change significantly increased CD8+ T cells in tumor and lymph, and also promoted the maturation of DCs, which effectively inhibited tumor growth and prolonged the survival of tumor-bearing mice.
Collectively, high affinity to albumin confers LsiRNA as a potent tumor treatment. Meanwhile, it also provides an idea for developing a new siRNA delivery system.
Declaration of competing interestY. Huang is the co-founder of Rigerna Therapeutics. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Beijing Natural Science Foundation (No. 7214302), the Natural Science Foundation of Guangdong Province (No. 2019A1515010776), the National Natural Science Foundation of China (Nos. 31901053, 32001008, 32171394), the Beijing-Tianjin-Hebei Basic Research Cooperation Project (No. 19JCZDJC64100), the Beijing Nova Program from Beijing Municipal Science & Technology Commission (No. Z201100006820005), the National Key Research & Development Program of China (Nos. 2021YFE0106900, 2021YFA1201000, 2021YFC2302400). We thank the Biological and Medical Engineering Core Facilities of Beijing Institute of Technology, and Analysis and Testing Center of Beijing Institute of Technology for supporting experimental equipment, and staffs for valuable help with technical support.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108210.
| [1] |
Y. Weng, H. Xiao, J. Zhang, et al., Biotechnol. Adv. 37 (2019) 801-825. DOI:10.1016/j.biotechadv.2019.04.012 |
| [2] |
A. Wittrup, J. Lieberman, Nat. Rev. Genet. 16 (2015) 543-552. DOI:10.1038/nrg3978 |
| [3] |
K. Li, M. Lu, X. Xia, et al., Chin. Chem. Lett. 32 (2021) 1010-1016. DOI:10.1016/j.cclet.2020.09.010 |
| [4] |
S. Guo, K. Li, B. Hu, et al., Exploration 1 (2021) 35-49. DOI:10.1002/exp.20210008 |
| [5] |
B. Hu, B. Li, K. Li, et al., Sci. Adv. 8 (2022) eabm1418. DOI:10.1126/sciadv.abm1418 |
| [6] |
M. Jayaraman, S.M. Ansell, B.L. Mui, et al., Angew. Chem. Int. Ed. 51 (2012) 8529-8533. DOI:10.1002/anie.201203263 |
| [7] |
E. Khvalevsky, R. Gabai, I.H. Rachmut, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 20723-20728. DOI:10.1073/pnas.1314307110 |
| [8] |
J. Zhou, Y. Wu, C. Wang, et al., Nano Lett. 16 (2016) 6916-6923. DOI:10.1021/acs.nanolett.6b02915 |
| [9] |
A. Ellert-Miklaszewska, N. Ochocka, M. Maleszewska, et al., Nanomedicine 14 (2019) 2441-2458. DOI:10.2217/nnm-2019-0176 |
| [10] |
J. Liu, C. Chen, T. Wei, et al., Exploration 1 (2021) 21-34. DOI:10.1002/EXP.20210003 |
| [11] |
S. Kamerkar, V.S. LeBleu, H. Sugimoto, et al., Nature 546 (2017) 498-503. DOI:10.1038/nature22341 |
| [12] |
Y. Huang, Q. Cheng, X. Jin, et al., Biomater. Sci. 4 (2016) 494-510. DOI:10.1039/C5BM00429B |
| [13] |
Y. Huang, X. Wang, W. Huang, et al., Sci. Rep. 5 (2015) 12458. DOI:10.1038/srep12458 |
| [14] |
S.S. Kim, C. Ye, P. Kumar, et al., Mol. Ther. 18 (2010) 993-1001. DOI:10.1038/mt.2010.27 |
| [15] |
P. Kumar, H. Wu, J.L. McBride, et al., Nature 448 (2007) 39-43. DOI:10.1038/nature05901 |
| [16] |
M. Li, C. Wang, Z. Di, et al., Angew. Chem. Int. Ed. 58 (2019) 1350-1354. DOI:10.1002/anie.201810735 |
| [17] |
B. Liu, F. Hu, J. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 8804-8808. DOI:10.1002/anie.201903417 |
| [18] |
Y. Liu, J. Zhang, Y. Guo, et al., Exploration 2 (2022) 20210172. DOI:10.1002/EXP.20210172 |
| [19] |
S. Guo, B. Liu, M. Zhang, et al., Chin. Chem. Lett. 32 (2021) 102-106. DOI:10.1016/j.cclet.2020.11.024 |
| [20] |
M. Caillaud, M. El Madani, L. Massaad-Massade, J. Control. Release 321 (2020) 616-628. DOI:10.1016/j.jconrel.2020.02.032 |
| [21] |
T.M. Allen, P.R. Cullis, Adv. Drug Deliv. Rev. 65 (2013) 36-48. DOI:10.1016/j.addr.2012.09.037 |
| [22] |
O. Khorev, D. Stokmaier, O. Schwardt, et al., Bioorg. Med. Chem. 16 (2008) 5216-5231. DOI:10.1016/j.bmc.2008.03.017 |
| [23] |
B. Hu, L. Zhong, Y. Weng, et al., Signal Transduct. Target. Ther. 5 (2020) 101. DOI:10.1038/s41392-020-0207-x |
| [24] |
K.M. Brown, J.K. Nair, M.M. Janas, et al., Nat. Biotechnol. 40 (2022) 1500-1508. DOI:10.1038/s41587-022-01334-x |
| [25] |
E. Neumann, E. Frei, D. Funk, et al., Expert Opin. Drug Deliv. 7 (2010) 915-925. DOI:10.1517/17425247.2010.498474 |
| [26] |
C. Ji, X. Guo, Nat. Rev. Endocrinol. 15 (2019) 731-743. DOI:10.1038/s41574-019-0260-0 |
| [27] |
E.N. Hoogenboezem, C.L. Duvall, Adv. Drug Deliv. Rev. 130 (2018) 73-89. DOI:10.1016/j.addr.2018.07.011 |
| [28] |
Y. Zhao, C. Cai, M. Liu, et al., Int. J. Biol. Macromol. 153 (2020) 873-882. DOI:10.1016/j.ijbiomac.2020.03.060 |
| [29] |
G. Wang, Z. Zhou, Z. Zhao, et al., ACS Nano 14 (2020) 4890-4904. DOI:10.1021/acsnano.0c00974 |
| [30] |
H. Dong, S.E. Strome, D.R. Salomao, et al., Nat. Med. 8 (2002) 793-800. DOI:10.1038/nm730 |
| [31] |
M.J. Butte, M.E. Keir, T.B. Phamduy, et al., Immunity 27 (2007) 111-122. DOI:10.1016/j.immuni.2007.05.016 |
| [32] |
J.J. Park, R. Omiya, Y. Matsumura, et al., Blood 116 (2010) 1291-1298. DOI:10.1182/blood-2010-01-265975 |
| [33] |
B. Hu, Y. Weng, X.H. Xia, et al., J. Gene Med. 21 (2019) e3097. DOI:10.1002/jgm.3097 |
| [34] |
R. Liu, C. Luo, Z. Pang, et al., Chin. Chem. Lett. 34 (2023) 107518. DOI:10.1016/j.cclet.2022.05.032 |
| [35] |
S. Wilhelm, A.J. Tavares, Q. Dai, et al., Nat. Rev. Mater. 1 (2016) 16014. DOI:10.1038/natrevmats.2016.14 |
| [36] |
F. Danhier, J. Control. Release 244 (2016) 108-121. DOI:10.1016/j.jconrel.2016.11.015 |
| [37] |
J.W. Nichols, Y.H. Bae, J. Control. Release 190 (2014) 451-464. DOI:10.1016/j.jconrel.2014.03.057 |
| [38] |
M.N. Kundranda, J. Niu, Drug Des. Dev. Ther. 9 (2015) 3767-3777. DOI:10.2147/DDDT.S88023 |
| [39] |
J. Hu, X. Yuan, F. Wang, et al., Chin. Chem. Lett. 32 (2021) 1341-1347. DOI:10.1016/j.cclet.2020.11.006 |
| [40] |
W. Song, Z. Tang, N. Shen, et al., J. Control. Release 231 (2016) 94-102. DOI:10.1016/j.jconrel.2016.02.039 |
| [41] |
W. Song, Z. Tang, D. Zhang, et al., Small 11 (2015) 3755-3761. DOI:10.1002/smll.201500324 |
| [42] |
L. Tang, X. Yang, Q. Yin, et al., Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 15344-15349. DOI:10.1073/pnas.1411499111 |
| [43] |
S. Qin, B.Z. Fite, M.K. Gagnon, et al., Ann. Biomed. Eng. 42 (2014) 280-298. DOI:10.1007/s10439-013-0895-2 |
| [44] |
Q. Sun, Z. Zhou, N. Qiu, et al., Adv. Mater. 29 (2017) 201606628. |