 2023, Vol. 34
2023, Vol. 34 


b School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, China
Depletion of fossil feedstocks and deterioration of environmental conditions have rapidly stimulated the development of renewable biomass resources and the sustainable production of fuels and chemicals [1-3]. Cellulose and hemicellulose, the main components of nonedible lignocellulosic biomass, can be converted into monosaccharide (glucose and xylose), enabling the production of high value-added chemicals, such as furfural, hydroxymethylfurfural, sugar alcohol, and especially sugar acid [4-8]. Gluconic acid and xylonic acid are both the global top thirty value-added chemicals from biomass, and have wide applications in the food, agriculture, medicine, and construction industries [9-11]. As a result, numerous studies have focused on monosaccharide oxidation into sugar acid [12-14]. However, due to the oxidative cleavage of C–C bonds in monosaccharide and the over-oxidation reaction of sugar acid, the development of green and efficient catalytic systems remains a challenge [15].
The selective conversion of monosaccharide into sugar acid requires the oxidation of aldehyde group into carboxyl group [14]. It is generally accepted that supported metal catalysts, such as Au, Ru, Pt, and Pd, have high catalytic activity and selectivity for the oxidation of aldehyde group to carboxyl group [16-18]. Particularly, Au-based catalysts are considered outstanding because of their low sensitivity to oxygen poisoning and catalytic activity over a broad pH range [19]. The catalyst support is also an important factor for its activity and stability. Carbon materials are widely used as catalyst supports due to their high stability and recyclability in water over a wide range of pH values [20]. However, since carbon supports are hydrophobic and nonpolar, surface functionalization is typically required during the preparation of the catalyst in order to improve the wetting properties of the carbon surface [21,22]. The carbon surface itself is modified by inserting other elements, such as oxygen and nitrogen, in order to introduce oxygen- or nitrogen-containing functional groups [23,24]. These functional groups determine the acidity or alkalinity of the carbon support and exert important effects on the size, distribution, and loading of supported metal while improving catalyst performance [25]. Jongh et al. investigated the effects of different carbon-based surface functional groups on the activity of Au catalysts for the oxidation of HMF [20]. The activity and stability of carbon-supported Au catalysts depend on the surface functional groups of the support. Surface groups of basic carbon contribute more substantially to the oxidation of HMF into FDCA than surface groups of acidic carbon. Nevertheless, the effect of surface functional groups on the oxidation of monosaccharide remains unclear, it is thus necessary to understand the role of surface functional groups in providing a basis for designing and developing promising catalysts for the oxidation of monosaccharide.
In this work, we demonstrate a surface-functionalized carbon nanotube-supported gold catalyst for base-free selective oxidation of monosaccharide into sugar acid. The surface of the catalyst successfully introduced oxygen- or nitrogen-containing functional groups. We catalyzed the oxidation of glucose and xylose in order to investigate the universality of the catalyst for the oxidation of monosaccharide. To understand the effect of different surface functional groups on monosaccharide oxidation, a kinetic model of monosaccharide oxidation into sugar acid was developed and its kinetic parameters are reported. For in-depth understand the effect of the functional groups on the surface of CNT supports and reveal the underlying reaction mechanism of selective oxidation of monosaccharide to sugar acid, a systematic DFT calculation was performed. Additionally, the catalyst was investigated in terms of its stability and recyclability.
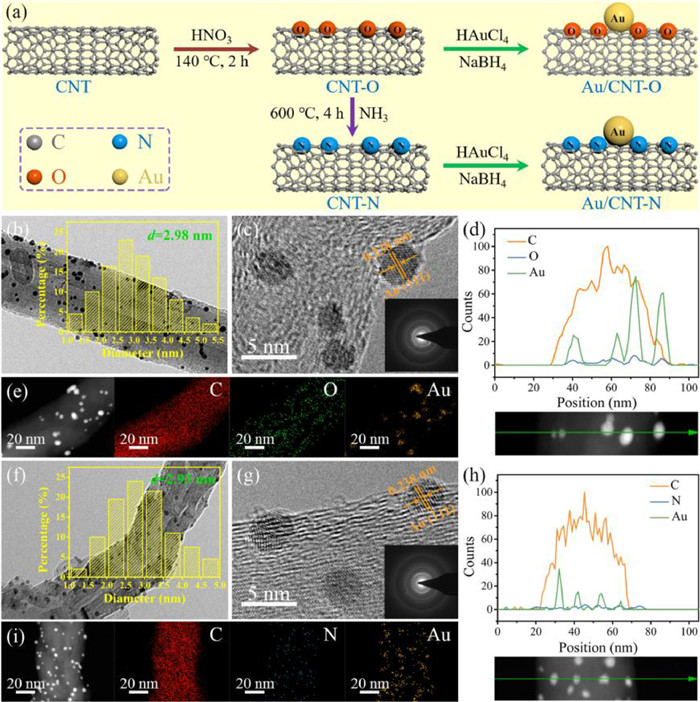
The synthesis of Au/CNT-O and Au/CNT-N is illustrated in Fig. 1a. First, using commercial CNT as the matrix, oxygen-containing functional groups were introduced at the CNT surface by reflux treatment in concentrated HNO3. Subsequently, oxygen-containing functional groups on CNT surface were replaced by nitrogen-containing functional groups using heat treatment with NH3. Finally, Au/CNT-O and Au/CNT-N were fabricated by immobilizing PVA-stabilized colloidal Au nanoparticles at the surfaces of CNT-O and CNT-N and by reduction with NaBH4.

|
Download:
|
| Fig. 1. (a) Illustration of the preparation of Au/CNT-O and Au/CNT-N; TEM, HR-TEM, line scan, and elemental mapping images of (b-e) Au/CNT-O, and (f-i) Au/CNT-N. | |
The surface morphology and structure of Au/CNT-O and Au/CNT-N were characterized by SEM (Fig. S1 in Supporting information) and TEM (Fig. 1). As shown in Fig. S1, Au/CNT-O and Au/CNT-N have similar surface morphology and structure, indicating that the pretreatments have no discernible effects. Figs. 1b and f show TEM images of Au/CNT-O and Au/CNT-N and distribution histograms of Au nanoparticle size. This demonstrates that Au nanoparticles were successfully immobilized and uniformly dispersed across the surface of the CNT-O and CNT-N supports. The average sizes of Au nanoparticles of Au/CNT-O and Au/CNT-N were 2.98 nm and 2.93 nm, respectively. No significant differences were observed in the average size of Au nanoparticles between Au/CNT-O and Au/CNT-N. This indicates that the pretreatments have no significant influence on the size of Au nanoparticles. HR-TEM images (Figs. 1c and g) clearly display lattices with a spacing of 0.238 nm, which was assigned to the (111) face of metallic Au [26]. The insets in Figs. 1c and g show the diffraction pattern of Au nanoparticles. In addition, a line scan of Au/CNT-O and Au/CNT-N was performed. As shown in Figs. 1d and h, this scan verified the tubular morphologies of Au/CNT-O and Au/CNT-N. Elemental mapping images (Figs. 1e and i, and Figs. S1b and d) also clearly demonstrate that C, O and Au were evenly distributed in Au/CNT-O while C, N and Au were evenly distributed in Au/CNT-N. The results of the line scan and elemental mapping suggest that oxygen-containing functional groups were successfully introduced into Au/CNT-O whereas nitrogen-containing functional groups were successfully introduced into Au/CNT-N.
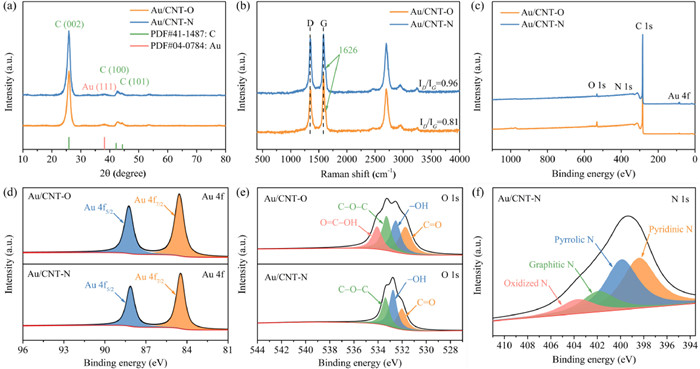
The XRD patterns of Au/CNT-O and Au/CNT-N (Fig. 2a) show series of characteristic reflections at 2θ values of 26.4°, 42.2°, and 44.4°, indexed to the (002), (100), and (101) facets of a typical graphitic carbon (JCPDS, PDF#41–1487), respectively [27,28]. Excepting the characteristic peaks of graphitic carbon, one weak peak is also located at approximately 38.3°, which can be assigned to the (111) facet of metallic Au (JCPDS, PDF#04–0784) [3,29]. This result confirms that Au nanoparticles were successfully immobilized on the surface of pretreated CNT supports. No obvious differences in crystal texture were observed between Au/CNT-O and Au/CNT-N in XRD patterns. The degree of graphitization was analyzed using Raman spectroscopy. For all carbon catalysts, the characteristic D and G bands occur at 1340 cm−1 and 1591 cm−1 and are indexed to disordered carbon atoms and sp2 hybridized graphitic carbon atoms, respectively [30,31]. As shown in Fig. 2b, the value of ID/IG of Au/CNT-N is 0.96, higher than Au/CNT-O, indicating the increased formation of edge and topological defects on the Au/CNT-N surface [32]. These edge and abundant defect sites can modulate and tune the electronic and surface properties of Au/CNT-N, thereby contributing to the oxidation of monosaccharide [33,34]. A weak peak located at 1626 cm−1 in Raman spectra is attributed to the characteristic peak of multiwalled carbon nanotube. Au/CNT-O and Au/CNT-N were further confirmed using FT-IR spectroscopy. As shown in Fig. S2 (Supporting information), peaks at 1154 and 3452 cm−1 are attributed to the characteristic stretching vibrations of C−O and C−OH bonds, respectively [35]. Notably, the peak at 1635 cm−1 could occur as a result of overlap among the C=C, C=N, and C=O bonds [36]. The peak intensity for C−O and C−OH of Au/CNT-O is higher than that of Au/CNT-N, while the peak intensity for C=N is lower than that of Au/CNT-N, confirming that Au/CNT-O successfully introduced oxygen-containing functional groups while Au/CNT-N successfully introduced nitrogen-containing functional groups.

|
Download:
|
| Fig. 2. (a) XRD patterns, (b) Raman spectra, (c) wide XPS survey spectra, high-resolution (d) Au 4f spectra, (e) O 1s spectra, and (f) N 1s spectra of Au/CNT-O and Au/CNT-N. | |
The chemical composition and bonding configurations of Au/CNT-O and Au/CNT-N surfaces were investigated using X-ray photoelectron spectroscopy (XPS). The presence of Au, C, and O in Au/CNT-O and Au, C, O and N elements in Au/CNT-N was observed in the wide XPS spectra (Fig. 2c). The corresponding atomic percentages of elements are summarized in Table S1 (Supporting information). The Au contents of Au/CNT-O and Au/CNT-N are 0.88% and 0.86%, respectively, slightly lower than theoretical values. The Au loadings of Au/CNT-O and Au/CNT-N were determined by ICP-MS. As shown in Table S1, the Au contents of Au/CNT-O and Au/CNT-N are close to XPS results, because CNT-O and CNT-N are hydrophobic carbon materials, and it is difficult to absorb Au nanoparticles onto the surfaces of CNT-O and CNT-N [36].
In order to obtain clear information on Au valence, high-resolution XPS spectra of Au 4f were obtained, the results of which are displayed in Fig. 2d. Two well-defined peaks at electron-binding energy of 87.9 and 84.2 eV were attributed to Au 4f7/2 and Au 4f5/2, respectively. This indicates that Au3+ on CNT-O and CNT-N support surfaces was successfully reduced to Au0 nanoparticles by NaBH4 reduction, in accordance with previous findings [12]. The O content of Au/CNT-O is 5.07%, further implying the introduction of various oxygen-containing functional groups. However, an obvious decrease in O content was observed after thermal treatment under NH3 occurs due to the degradation of a part of the thermally unstable oxygen-containing functional groups [37]. The fitted high-resolution O 1s spectra of Au/CNT-O and Au/CNT-N for studying oxygen-containing functional groups are shown in Fig. 2e. The O 1s peaks of Au/CNT-O at electron-binding energy of 531.8, 532.7, 533.5 and 534.4 eV are attributed to oxygen in C=O, −OH, C−O−C, and O=C−OH, respectively [38,39]. Compared with Au/CNT-O, the oxygen-containing functional groups of Au/CNT-N consist of C=O, −OH, and C−O−C without the O=C−OH group. Furthermore, no O=C−OH group is present in fitted high-resolution C 1s spectra of Au/CNT-N (Fig. S3 in Supporting information). These results indicate that some oxygen-containing functional groups were lost following the amination treatment. Since N content of Au/CNT-N is 1.03%, we confirm that some of these oxygen-containing functional groups were replaced with nitrogen-containing functional groups. Fig. 2f displays the high-resolution XPS spectra of N 1s. Four peaks at 398.4, 399.9, 401.9 and 403.7 eV are attributed to pyridinic N, pyrrolic N, graphitic N and oxidized N, respectively [40]. It is generally accepted that the pyridinic and pyrrolic N play a vital role in catalytic oxidation reaction [41].
N2 adsorption-desorption was employed to analyze the surface area and porous structure of Au/CNT-O and Au/CNT-N. As shown in Fig. S4 (Supporting information), the N2 sorption isotherms of Au/CNT-O and Au/CNT-N exhibit type Ⅳ isotherm patterns, confirming the existence of abundant mesopores. The specific surface area (BET) and pore volume of Au/CNT-N are 118.18 m2/g and 0.43 m3/g, respectively, close to those of Au/CNT-O. This indicates that the surface area and porous structure of CNT-O and CNT-N did not undergo significant changes after amination. The corresponding pore size distribution curves indicate that Au/CNT-O and Au/CNT-N possess large pore volumes and are dominated by mesopores (Fig. S4 and Table S1 in Supporting information). The mean pore sizes of Au/CNT-O and Au/CNT-N are similar, being measured at 6.48 and 6.58 nm, respectively. The existence of mesopores provides a high density of active sites and is conducive to the transportation of reaction intermediates [40]. The acid-base properties of Au/CNT-O and Au/CNT-N were studied using acid-base titration. The results for the densities of acid and base sites are summarized in Table 1. The initial pH values indicate that Au/CNT-O is acidic whereas Au/CNT-N is basic. The strong acid sites and total acid sites of Au/CNT-O account for 134.2 and 298.6 mmol/kg, respectively. The strong acidity of Au/CNT-O is generally attributed to oxygen-containing functional groups [20]. The total base sites of Au/CNT-N account for 8.2 mmol/kg. The weak basicity of Au/CNT-N is derived from nitrogen-containing functional groups [20]. In summary, Au/CNT-O and Au/CNT-N have similar morphologies, structures, and Au loading but differ in their surface properties, including surface elemental composition, the nature and concentration of surface functional groups, and acid-base properties. These different surface properties exert important effects on the catalytic performance of Au/CNT-O and Au/CNT-N.
|
|
Table 1 Acid-base properties of Au/CNT-O and Au/CNT-N. |

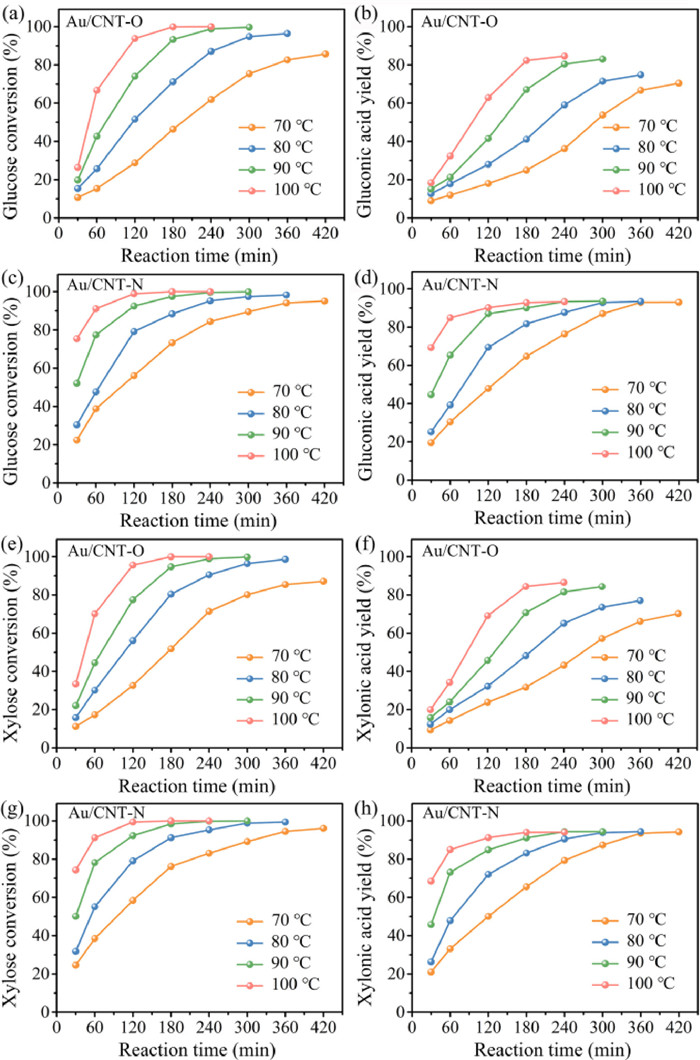
Monosaccharide oxidation experiments were performed at fixed reaction conditions of 1 mmol glucose or xylose in 30 mL water, 100 mg catalyst, and 0.5 MPa O2. The effect of temperature on glucose oxidation over Au/CNT-O or Au/CNT-N is illustrated in Figs. 3a–d. Analysis of these results indicated that the oxidation of glucose into gluconic acid occurs in the kinetic-limited regime. Glucose conversion in the Au/CNT-O and Au/CNT-N systems varied from 10.7% to 100% and 22.6% to 100%, respectively. Reaction temperature has a significant effect on glucose oxidation. Indeed, as can be seen that elevated temperatures result in higher reaction rates. For instance, the reaction time required to reach the highest glucose conversion was 420 min at 70 ℃ but only 180 min at 100 ℃ when using either Au/CNT-O or Au/CNT-N as the catalyst. In addition, increasing the reaction temperatures enhanced the gluconic acid yield. The gluconic acid yields in the Au/CNT-O and Au/CNT-N systems were 80.4% and 93.3%, respectively; these were obtained in 240 min at 90 ℃. When the reaction temperature was increased to 100 ℃, the gluconic acid yield was not significantly enhanced. Furthermore, irrespective of temperature, increasing the reaction time did not influence the gluconic acid yield, suggesting that gluconic acid is stable under such conditions and that secondary degradation of gluconic acid did not occur in either of the tested systems.

|
Download:
|
| Fig. 3. Effect of reaction temperature on glucose and xylose conversion, and gluconic acid and xylonic acid yield by Au/CNT-O (a, b and e, f) or Au/CNT-N (c, d and g, h). | |
The glucose conversion and gluconic acid yield in the Au/CNT-O system were both noticeably lower than those in the Au/CNT-N system under the same reaction temperature and time. The highest gluconic acid yield in the Au/CNT-N system (93.3%) was higher than that in the Au/CNT-O system (84.6%), indicating that Au/CNT-N is more conducive to glucose oxidation into gluconic acid than Au/CNT-O. Observed differences in catalytic performance are due to differences between the surface functional groups of Au/CNT-O and Au/CNT-N [20]. Therefore, we conclude that nitrogen-containing functional groups in Au/CNT-N are more favorable to the selective oxidation of glucose into gluconic acid than oxygen-containing functional groups in Au/CNT-O.
In order to verify the universality of these conclusions, a similar set of experiments was performed by using xylose as the reactant; the results of these experiments are shown in Figs. 3e–h. The tendency of xylose oxidation was found to be similar with that of glucose oxidation. Whether in the Au/CNT-O or Au/CNT-N system, xylose oxidation into xylonic acid is also considered to be in the kinetic-limited regime, and the reaction temperature has a significant effect. Xylose conversion and xylonic acid yield both increased with increasing reaction temperature and reaction time. Xylose conversion in the Au/CNT-O and Au/CNT-N systems varied from 11.2% to 100% and 24.6% to 100%, respectively. When the reaction temperature was increased from 90 ℃ to 100 ℃, the rection time required to obtain the maximum xylonic acid yield of decreased from 300 min to 240 min, irrespective of the system used. The highest xylonic acid yields in the Au/CNT-O and Au/CNT-N systems were 84.5% and 94.3%, respectively. Xylonic acid is also stable under the experimental conditions. Likewise, the xylose conversion and xylonic acid yield in the Au/CNT-N system were both higher than that in the Au/CNT-O system under the same reaction conditions, which further suggests that nitrogen-containing functional groups are more beneficial to selective oxidation of xylose into xylonic acid than oxygen-containing functional groups. Thus, our conclusions are generally applicable to the oxidation of monosaccharide.
The stability and recyclability of Au/CNT-N were investigated by reusing the catalyst in consecutive catalytic runs. In order to remove adsorbed substances, the recovered catalyst was treated at 300 ℃ for 1 h in a 10% H2/Ar atmosphere [12]. Fig. S5 (Supporting information) shows the conversion and yield of five subsequent runs at 90 ℃ for 240 min, showing that the Au/CNT-N is remarkably stable and recyclable. Glucose and xylose conversion, as well as gluconic acid and xylonic acid yield, did not show significant decline after five cycle runs. Hence, Au/CNT-N is stable in the monosaccharide oxidation reaction system. Additionally, the real active centers of catalysts are the Au nanoparticles (Table S2 in Supporting information).
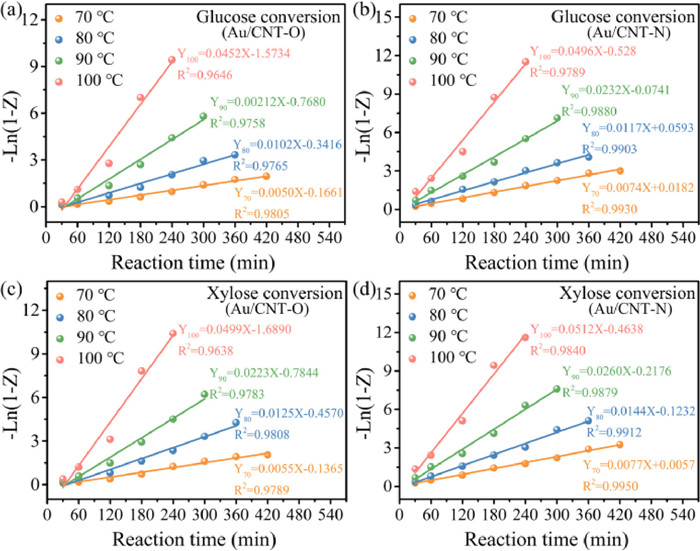
The overall reaction network used for the development of the kinetic model of monosaccharide oxidation is displayed in Fig. S6 (Supporting information). The oxidation of monosaccharide can be divided into two key steps [12,19]: (1) monosaccharide oxidation to produce sugar acid, and (2) monosaccharide decomposition to generate byproducts. Fig. 4 shows −Ln(1 − Z) versus reaction time for glucose and xylose conversion under different reaction temperatures. Their high linearity confirmed the first-order dependence of glucose and xylose decomposition, indicating the validity of the constructed kinetic model, which ensures that the calculated reaction rate constants and activation energy are reasonable.

|
Download:
|
| Fig. 4. −Ln(1 − Z) versus reaction time for glucose or xylose conversion base on different reaction temperature over Au/CNT-O (a, c) or Au/CNT-N (b, d). | |
Table 2 summarizes the effects of reaction temperature on the reaction rate constants for oxidation of glucose or xylose using Au/CNT-O or Au/CNT-N catalysts. As shown in Table 2, whether using an Au/CNT-O or Au/CNT-N catalyst, the decompositon rate constants of glucose (kG) and the formation rate constants of gluconic acid (kG1) increased with increasing reaction temperature, which confirms that increasing the reaction temperature is conducive to accelerating glucose decomposition and gluconic acid formation. Nevertheless, the formation rate constants of gluconic acid (kG1) were always found to be lower than those of glucose (kG) at any temperature, suggesting that only part of the glucose was oxidized to gluconic acid. Thus, the theoretical yield of gluconic acid cannot be obtained from the glucose oxidation reaction. In addition, the formation rate constants of gluconic acid (kG1) were always higher than those of byproducts (kG2), indicating that gluconic acid is the main product of glucose oxidation under the experimental conditions used.
|
|
Table 2 Reaction rate constant for oxidation of glucose or xylose using Au/CNT-O or Au/CNT-N catalysts. |
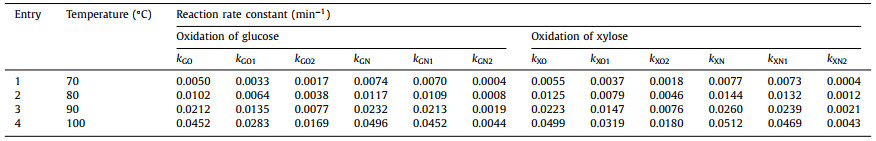
On the other hand, the rate constants of glucose decompositon (kGN) and gluconic acid formation (kGN1) using Au/CNT-N as catalyst were both larger than the corresponding rate constants using Au/CNT-O as the catalyst under similar reaction temperatures, indicating that Au/CNT-N accelerates the decomposition of glucose and the formation of gluconic acid to a greater degree than Au/CNT-O. Conversely, the higher rate constants of byproduct formation (kGO2) using Au/CNT-O as catalyst compared to those for kGN2 with Au/CNT-N as the catalyst implies that byproducts are form more easier in the Au/CNT-O system than in the Au/CNT-N system.
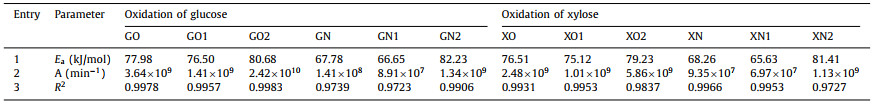
Table 3 also provides the activation energy and pre-exponential factor for oxidation of glucose or xylose using Au/CNT-O or Au/CNT-N catalysts. These constants (Ea-G and AG) were obtained from the slopes and intercepts of the fitted straight lines in Fig. S7 (Supporting information). The activation energy (Ea-G2) of glucose decomposition to byproducts is higher than that of glucose oxidation to gluconic acid (Ea-G1), which suggests that high temperatures are not favorable for gluconic acid formation; this is likely a result of byproduct formation. Hence, the highest gluconic acid yields were not achieved at the highest temperatures (Figs. 3b and d). Additionally, the activation energies of glucose decomposition (Ea-GO = 77.98 kJ/mol) and gluconic acid formation (Ea-GO1 = 76.50 kJ/mol) using an Au/CNT-O catalyst were both higher than the corresponding activation energies (Ea-GN = 67.78 kJ/mol and Ea-GN1 = 66.65 kJ/mol) using Au/CNT-N as a catalyst, whereas the activation energy of byproduct formation using Au/CNT-O as catalyst were lower than those when using Au/CNT-N as the catalyst. This indicates that the consumed energy resulting from glucose oxidation into gluconic acid in the Au/CNT-N system is lower than that of the Au/CNT-O system, and side reactions are relatively inhibited. In summary, Au/CNT-N is more conducive to the oxidation of glucose into gluconic acid than Au/CNT-O. Differences in reaction rate constants and activation energy between the Au/CNT-O and Au/CNT-N reaction systems are attributed to their different surface functional groups. The nitrogen-containing functional groups in Au/CNT-N accelerate glucose oxidation and enhance gluconic acid selectivity to a greater extent than the oxygen-containing functional groups in Au/CNT-O.
|
|
Table 3 Activation energy and pre-exponential factor for oxidation of glucose or xylose using Au/CNT-O or Au/CNT-N catalysts. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
As shown in Table 2, the regularity of xylose oxidation is similar to that of glucose oxidation. The rate constants of xylose decomposition (kX) and xylonic acid formation (kX1) both increased with increasing reaction temperature. The rate constants of xylonic acid formation (kX1) were higher than those of byproducts formation (kX2) at any temperature, confirming that xylonic acid is the main product of xylose oxidation under the tested experimental conditions.
The values of the activation energy (Ea-X) and pre-exponential factor (AXE) were determined from the Arrhenius plots in Fig. S8 (Supporting information). As shown in Table 3, the conversion of xylose to byproducts had the highest activation energy, indicating that high temperature enhances byproduct formation. Similarly, the activation energies of xylose decomposition (Ea-XO = 76.51 kJ/mol) and xylonic acid formation (Ea-XO1 = 75.12 kJ/mol) in the Au/CNT-O system were also higher than the corresponding activation energies (Ea-XN = 68.26 kJ/mol and Ea-XN1 = 65.63 kJ/mol) in the Au/CNT-N system, suggesting that nitrogen-containing functional groups are more favorable to selective oxidation of xylose into xylonic acid than oxygen-containing functional groups. This further verifies that the conclusions are generally applicable to monosaccharide oxidation into sugar acid.
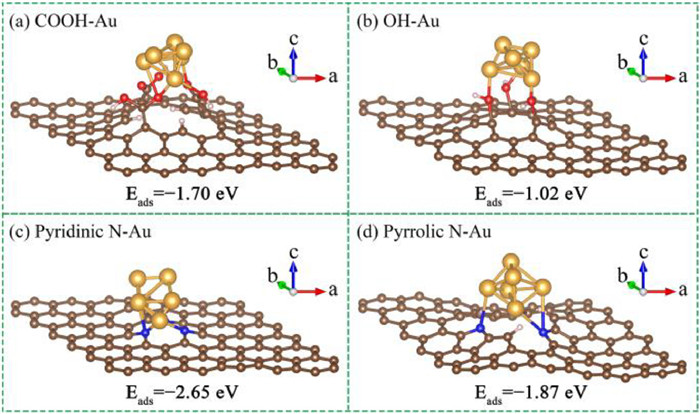
In order to in-depth understand the effect of the functional groups on the surface of CNT supports and reveal the underlying reaction mechanism of selective oxidation of monosaccharide to sugar acid, a systematic DFT calculation was carried out. According to the XPS results (Figs. 2e and f), the acidic oxygen-containing functional groups of CNT-O include carboxyl and hydroxy groups, while the basic nitrogen-containing functional groups of CNT-N are attributed to pyridinic N and pyrrolic N atoms. The structures of a graphene layer functionalized by three carboxyl groups, three hydroxy groups, three pyridinic N atoms, and three pyrrolic N atoms are displayed in Fig. S9 (Supporting information). The DFT calculation allows us to separately predict the adsorption of a gold cluster on functional groups under the stable configuration, which is hard to be experimentally studied because of the difficulty of preparing carbon materials containing a single type of functional groups [42,43]. Therefore, the optimum structures of Au6 cluster stabilized by oxygen-containing or nitrogen-containing functional groups on a graphene layer were studied.
Fig. 5 demonstrates the optimum structures of Au6 cluster on a graphene layer doped by a vacant site with oxygen-containing or nitrogen-containing functional groups, and the adsorption energy of Au6 cluster is summarized in Table S1. As shown in Figs. 5a and b (Au/CNT-O catalyst), the Au6 cluster was chemically bonded with and stabilized by three carboxyl groups or three hydroxy groups. The adsorption energy of Au6 cluster on carboxyl groups is −1.70 eV, which is higher than that on hydroxy groups (−1.02 eV), suggesting that the carboxyl groups are more conducive to immobilizing Au on CNT-O support. For Au/CNT-N catalyst (Figs. 5c and d), the Au6 cluster was adsorbed on three pyridinic N atoms or three pyrrolic N atoms, and the corresponding adsorption energies were −2.65 eV and −1.87 eV, respectively, which indicates that the adsorption of Au6 cluster on the pyridinic N atoms is stronger than that on the pyrrolic N atoms and the pyridinic N atoms play a more important role in immobilization of Au on CNT-N support. Hence, the theoretically optimal structures of Au/CNT-O and Au/CNT-N were the Au6 cluster on a graphene layer doped by a vacant site with three carboxyl groups and three pyridinic N atoms, respectively.

|
Download:
|
| Fig. 5. The geometric models of (a, b) Au/CNT-O and (c, d) Au/CNT-N catalyst. | |
{kind=link}
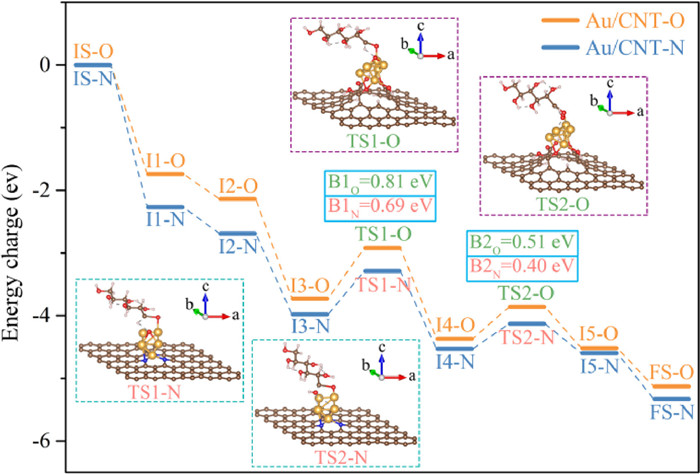
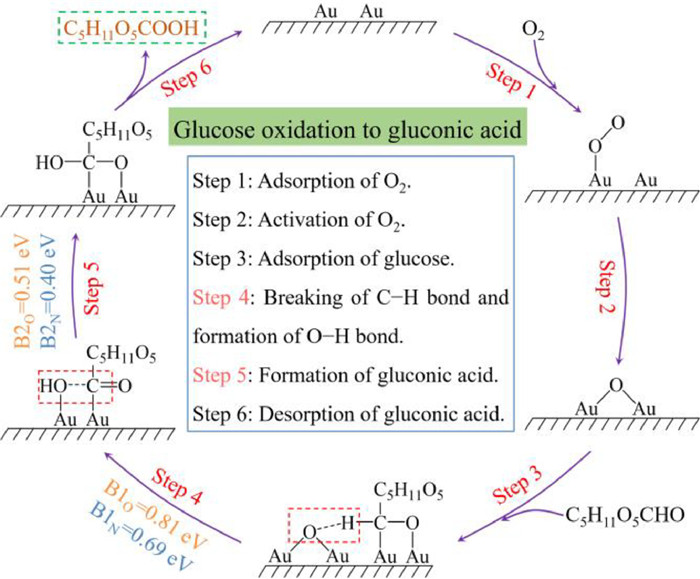
In addition, the reaction mechanism of monosaccharide oxidation into sugar acid was researched using glucose oxidation as an example. For comprehensively evaluate the catalytic oxidation of glucose into gluconic acid, the DFT calculation was performed on the theoretically optimal structures of Au/CNT-O or Au/CNT-N. The reaction energy profile and the structures of the intermediates and transition states for the glucose oxidation into gluconic acid on the theoretically optimal structures of Au/CNT-O or Au/CNT-N are exhibited in Fig. 6, Figs. S10 and S11 (Supporting information). The O2 molecule is first adsorbed on the Au6 cluster (I1-O and I1-N) and then is activated to generate the chemisorbed oxygen (I2-O and I2-N). Afterwards, the glucose molecule is adsorbed on the Au6 cluster (I3-O and I3-N) and the corresponding adsorption energies on Au/CNT-O and Au/CNT-N are −1.29 eV and −1.59 eV, respectively, implying strong binding of glucose on the Au6 cluster. After the adsorption of glucose, the formyl C−H bond is broken by the chemisorbed oxygen to form dehydrogenated glucose (I4-O and I4-N). Meanwhile, the activated OH is generated by transferring H from the formyl C−H bond to chemisorbed oxygen. TS1-O and TS1-N are the transition states in the activation of formyl C−H bond and the formation of activated OH. The activation energy barriers for C−H dissociation and activated OH generation over Au/CNT-O and Au/CNT-N are 0.81 eV and 0.69 eV, respectively. Then, the dehydrogenated glucose combines with the activated OH to form the chemisorbed gluconic acid (I5-O and I5-N), and the energy barriers over Au/CNT-O and Au/CNT-N are 0.51 eV (TS2-O) and 0.40 eV (TS2-N). Finally, the chemisorbed gluconic acid desorbs from the Au6 cluster to generate a gluconic acid molecule.

|
Download:
|
| Fig. 6. Energy profile of glucose oxidation into gluconic acid over Au/CNT-O or Au/CNT-N. | |
{kind=link}
As displayed in Fig. 7, the reaction pathway of glucose oxidation into gluconic acid over Au/CNT-O or Au/CNT-N involves the adsorption of O2, activation of O2, adsorption of glucose, dissociation of the formyl C−H bond and formation of O−H bond, formation of chemisorbed gluconic acid, and desorption of gluconic acid. The key steps during the glucose oxidation into gluconic acid are the dissociation of the formyl C−H bond and formation of O−H bond, and formation of chemisorbed gluconic acid [44]. The activation energy barriers for the glucose oxidation into gluconic acid over the Au/CNT-O and Au/CNT-N are summarized in Table S2. When Au/CNT-O is used as catalyst, the activation energy barriers for the dissociation of the formyl C−H bond and formation of O−H bond, and formation of chemisorbed gluconic acid are 0.81 eV and 0.51 eV, respectively. In the case of Au/CNT-N, the activation energy barriers for the dissociation of the formyl C−H bond and formation of O−H bond, and formation of chemisorbed gluconic acid are 0.69 eV and 0.40 eV, respectively, which are lower than that of Au/CNT-O, suggesting that it is easier to realize C−H dissociation and O−H generation, and form chemisorbed gluconic acid using Au/CNT-N as catalyst. This result indicates that nitrogen-containing functional groups are more favorable to selective oxidation of monosaccharide into sugar acid than oxygen-containing functional groups, which is in good agreement with results of experiment and kinetic study.

|
Download:
|
| Fig. 7. The reaction pathway of glucose oxidation into gluconic acid over Au/CNT-O or Au/CNT-N. | |
{kind=link}
In conclusion, Au/CNT-O and Au/CNT-N are similar exception of their surface properties. Au/CNT-O introduces oxygen-containing functional groups whereas Au/CNT-N introduces nitrogen-containing functional groups. These different surface functional groups have an important effect on monosaccharide base-free oxidation to sugar acid. Nitrogen-containing functional groups are more favorable to the selective oxidation of monosaccharide into sugar acid than their oxygen-containing counterparts. The highest gluconic acid and xylonic acid yields of 93.3% and 94.3%, respectively, were obtained using an Au/CNT-N catalyst at 90 ℃ for 240 min under base-free conditions, which is higher than that obtained using an Au/CNT-O catalyst. Furthermore, Au/CNT-N catalyst shows remarkable stability and recyclability. The reaction model developed to study the kinetics of monosaccharide oxidation into sugar acid is pseudo homogeneous with good correlation between experimental and model data. The reaction rate constants for monosaccharide decomposition and sugar acid formation both increase with increasing reaction temperature. Additionally, the rate constants for monosaccharide decomposition and sugar acid formation in the Au/CNT-N system are higher than those in the Au/CNT-O system, while the activation energies of monosaccharide decomposition and sugar acid formation are lower. DFT calculation revealed that the mechanism of glucose oxidation to gluconic acid involves the adsorption of O2, activation of O2, adsorption of glucose, dissociation of the formyl C−H bond and formation of O−H bond, formation of chemisorbed gluconic acid, and desorption of gluconic acid. The key steps in the glucose oxidation are the dissociation of the formyl C−H bond and formation of O−H bond, and formation of chemisorbed gluconic acid. The activation energy barrier for the glucose oxidation over Au/CNT-N is lower than that of Au/CNT-O. The nitrogen-containing functional groups are more conducive to accelerating monosaccharide oxidation and enhancing sugar acid selectivity than oxygen-containing functional groups.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Youth Talent Support Program, National Natural Science Foundation of China (No. 31971614), State Key Laboratory of Pulp and Paper Engineering (No. 2022PY02), the National Program for Support of Topnotch Young Professionals (No. x2qsA4210090), Guangdong Basic and Applied Basic Research Foundation (Nos. 2021A1515110205, 2021A1515110622, 2021A1515110245 and 2020A1515110705), Science and Technology Basic Resources Investigation Program of China (No. 2019FY100900), the National Key Research and Development Program of China (No. 2021YFC2101604).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108370.
| [1] |
M. Wang, H.S. Tsai, C. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2807-2816. DOI:10.1016/j.cclet.2021.11.060 |
| [2] |
S. Guo, Q. Fang, Z. Li, et al., Nanoscale 11 (2019) 1326-1334. DOI:10.1039/c8nr08143c |
| [3] |
X. He, Green Energy Environ. 6 (2021) 15-21. DOI:10.1016/j.gee.2020.04.003 |
| [4] |
M.V. Morales, J.M. Conesa, A. Guerrero-Ruiz, et al., Carbon 182 (2021) 265-275. DOI:10.1016/j.carbon.2021.06.007 |
| [5] |
L. Zhang, Y. Tian, Y. Wang, et al., Chin. Chem. Lett. 32 (2021) 2233-2238. DOI:10.1016/j.cclet.2020.12.030 |
| [6] |
X. Guo, F. Guo, Y. Li, et al., Appl. Catal. A: Gen. 558 (2018) 18-25. DOI:10.1016/j.apcata.2018.03.027 |
| [7] |
M. Sun, Y. Wang, C. Sun, et al., Chin. Chem. Lett. 33 (2022) 385-389. DOI:10.1016/j.cclet.2021.05.009 |
| [8] |
A. Delparish, A. Uslu, Y. Cao, et al., Chem. Eng. J. 438 (2022) 135393. DOI:10.1016/j.cej.2022.135393 |
| [9] |
W. Niu, M.N. Molefe, J.W. Frost, J. Am. Chem. Soc. 125 (2003) 12998-12999. DOI:10.1021/ja036391+ |
| [10] |
I. Witońska, M. Frajtak, S. Karski, Appl. Catal. A: Gen. 401 (2011) 73-82. DOI:10.1016/j.apcata.2011.04.046 |
| [11] |
J. Ma, L. Zhong, X. Peng, et al., Green Chem. 18 (2016) 1738-1750. DOI:10.1039/C5GC01727K |
| [12] |
X. Meng, Z. Li, D. Li, et al., Green Chem. 22 (2020) 2588-2597. DOI:10.1039/c9gc04333k |
| [13] |
T. Rafaïdeen, S. Baranton, C. Coutanceau, Appl. Catal. B: Environ. 243 (2019) 641-656. DOI:10.1016/j.apcatb.2018.11.006 |
| [14] |
Z. Li, D. Li, J. Ma, et al., Renew. Energy 175 (2021) 650-659. DOI:10.3390/met11040650 |
| [15] |
J. Yin, Q. Zhang, C. Yang, et al., Catal. Sci. Technol. 10 (2020) 2231-2241. DOI:10.1039/c9cy02393c |
| [16] |
M. Jing, W. Song, Y. Li, et al., Chem. Eng. J. 433 (2022) 133599. DOI:10.1016/j.cej.2021.133599 |
| [17] |
X. Zhu, Z. Hu, M. Huang, et al., Chin. Chem. Lett. 32 (2021) 2033-2037. DOI:10.1016/j.cclet.2020.11.071 |
| [18] |
N. Mei, B. Liu, J. Zheng, et al., Catal. Sci. Technol. 5 (2015) 3194-3202. DOI:10.1039/C4CY01407C |
| [19] |
P. Qi, S. Chen, J. Chen, et al., ACS Catal. 5 (2015) 2659-2670. DOI:10.1021/cs502093b |
| [20] |
B. Donoeva, N. Masoud, P.E. de Jongh, ACS Catal. 7 (2017) 4581-4591. DOI:10.1021/acscatal.7b00829 |
| [21] |
G. Goncalves, P.A.A.P. Marques, C.M. Granadeiro, et al., Chem. Mater. 21 (2009) 4796-4802. DOI:10.1021/cm901052s |
| [22] |
C. Ku, H. Guo, K. Li, et al., Chin. Chem. Lett. 34 (2023) 107298. DOI:10.1016/j.cclet.2022.03.021 |
| [23] |
R. Arrigo, M. Havecker, S. Wrabetz, et al., J. Am. Chem. Soc. 132 (2010) 9616-9630. DOI:10.1021/ja910169v |
| [24] |
W. Xia, C. Jin, S. Kundu, et al., Carbon 47 (2009) 919-922. DOI:10.1016/j.carbon.2008.12.026 |
| [25] |
A. Villa, M. Schiavoni, L. Prati, Catal. Sci. Technol. 2 (2012) 673-682. DOI:10.1039/c2cy00355d |
| [26] |
J. Ma, Z. Liu, J. Song, et al., Green Chem. 20 (2018) 5188-5195. DOI:10.1039/c8gc02618a |
| [27] |
X. Yang, H. Yu, F. Peng, et al., ChemSusChem 5 (2012) 1213-1217. DOI:10.1002/cssc.201100807 |
| [28] |
J. Wang, Z. Wei, Y. Gong, et al., Chem. Commun. 51 (2015) 12859-12862. DOI:10.1039/C5CC02593A |
| [29] |
C. Megías-Sayago, K. Chakarova, A. Penkova, et al., ACS Catal. 8 (2018) 11154-11164. DOI:10.1021/acscatal.8b02522 |
| [30] |
S. Zhao, Y. Cheng, J. Veder, et al., ACS Appl. Energy Mater. 1 (2018) 5286-5297. |
| [31] |
D. Bégin, G. Ulrich, J. Amadou, et al., J. Mol. Catal. A: Chem. 302 (2009) 119-123. DOI:10.1016/j.molcata.2008.12.006 |
| [32] |
Y. Jia, L. Zhang, A. Du, et al., Adv. Mater. 28 (2016) 9532-9538. DOI:10.1002/adma.201602912 |
| [33] |
C. Tang, Q. Zhang, Adv. Mater. 29 (2017) 1604103. DOI:10.1002/adma.201604103 |
| [34] |
Y. Ma, S. Jiang, G. Jian, et al., Energy Environ. Sci. 2 (2009) 224-229. DOI:10.1039/B807213M |
| [35] |
Z. Zhang, M. Dou, H. Liu, et al., Small 12 (2016) 4193-4199. DOI:10.1002/smll.201601617 |
| [36] |
T.O. Eschemann, W.S. Lamme, R.L. Manchester, et al., J. Catal. 328 (2015) 130-138. DOI:10.1016/j.jcat.2014.12.010 |
| [37] |
J.L. Figueiredo, J. Mater. Chem. A 1 (2013) 9351-9364. DOI:10.1039/c3ta10876g |
| [38] |
H. Schmiers, J. Friebel, P. Streubel, et al., Carbon 37 (1999) 1965-1978. DOI:10.1016/S0008-6223(99)00071-8 |
| [39] |
C. Hu, L. Dai, Adv. Mater. 29 (2017) 1604942. DOI:10.1002/adma.201604942 |
| [40] |
H. Jiang, J. Gu, X. Zheng, et al., Energy Environ. Sci. 12 (2019) 322-333. DOI:10.1039/c8ee03276a |
| [41] |
X. Han, C. Li, Y. Guo, et al., Appl. Catal. A: Gen. 526 (2016) 1-8. DOI:10.5414/CN108701 |
| [42] |
Z. He, B. Dong, W. Wang, et al., ACS Catal. 9 (2019) 2893-2901. DOI:10.1021/acscatal.8b03965 |
| [43] |
D. Guo, R. Shibuya, C. Akiba, et al., Science 351 (2016) 361-365. DOI:10.1126/science.aad0832 |
| [44] |
P.N. Amaniampong, Q.T. Trinh, B. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 8928-8933. DOI:10.1002/anie.201503916 |