 2023, Vol. 34
2023, Vol. 34 


b Shanghai Skin Disease Hospital, Tongji University School of Medicine, Shanghai 200443, China;
c Fudan Zhangjiang Institute, Shanghai 201203, China;
d Center for Medical Research and Innovation, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai 201399, China
Peptide and protein drugs have shown great potential for decades owing to their low toxicity and high specific interactions with biological targets. In addition to vaccination and diagnosis, the main applications of peptide and protein drugs are treating chronic diseases, autoimmune disorders, AIDS as well as cancers [1]. However, their therapeutic efficiency is always hampered as most peptide and protein drugs exhibit short half-lives and rapid clearance from systematic circulation. Additionally, the abundant proteases and pH fluctuation in the metabolic system may confer denaturation or degradation of proteins, which subsequently induces severe immune responses [2].
The emergence of controlled and sustained drug delivery systems has revolutionized the pharmaceutical, especially for pharmaceuticals with short half-lives. By prolonging the drug release period, patient compliance has been largely improved by avoiding repeated dosing while guaranteeing therapeutic efficiency. Due to their low absorption and high enzymatic degradation of peptides and proteins, oral, pulmonary, nasal, ocular and transdermal administration were actually not commonly utilized in long-acting drug delivery [3]. In contrast, parenteral administrations including injection, implantation, and infusion are preferred as they could enable an excellent bioavailability and low first-pass elimination of peptides and proteins.
For long-acting delivery system, reservoirs based on versatile polymers have been studied for more than 50 years. One of the most popular polymers investigated is poly(lactic-co-glycolic acid) (PLGA), which has been approved by U.S. Food and Drug Administration (FDA) for clinical use and drug delivery owing to its superior biocompatibility and biodegradability [4,5]. PLGA-based implants not only provide a long-acting delivery of peptides and proteins, but also efficiently protect them from the harsh environment in vivo, such as abundant enzymes and unfavorable pH. The first development of PLGA-based products can be traced back to 1986, when the injectable suspension of PLGA microspheres were approved for prostate cancer treatment [6]. Afterwards, dozens of products based on PLGA have reached clinical trials or even the market. It is noteworthy that marketed PLGA implants are not merely developed for delivery of peptides and proteins, small molecular chemicals, such as risperidone and dexamethasone, are also formulated for long-acting therapy.
In this review, we aim to introduce the fundamental properties of PLGA, and summarize the current status of PLGA implants, fabrication technologies of peptides/proteins-loaded PLGA devices, as well as factors influencing the drug release processes. We hope this review can help to encourage and inspire formulation development and optimization for sustained delivery of protein drugs.
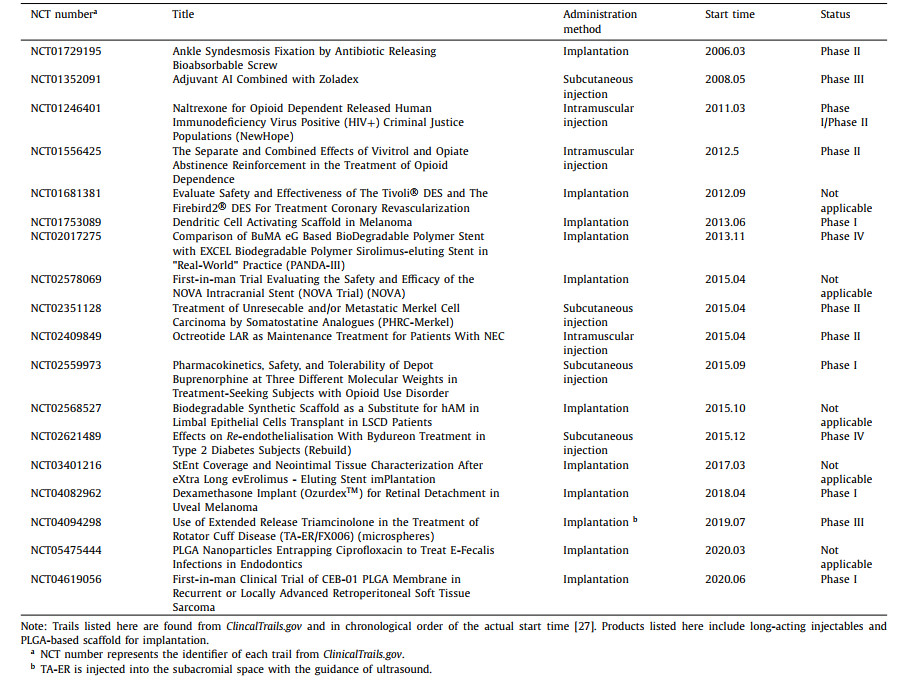
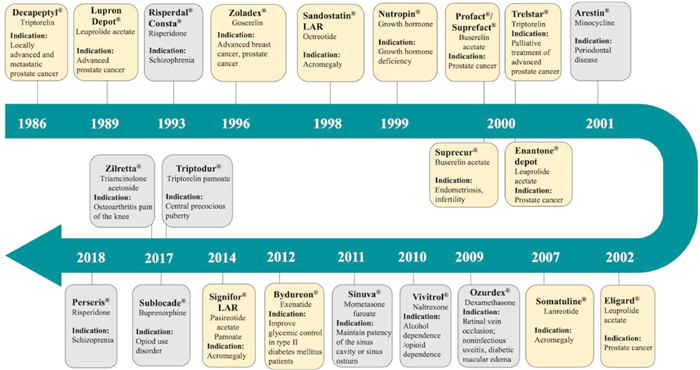
3. PLGA-based implants reaching clinical trials or marketAs PLGA has shown excellent safety and great potential to provide sustained drug release for drug products and tissue engineering scaffolds, more than twenty PLGA-based products have been approved by FDA since 1986, including microparticles, injectable in-situ hydrogels, and surgical solid implants [24]. Fig. 2 presents the approval timeline of implanted PLGA products for drug delivery in recent thirty years [6,25,26]. Decapeptyl® was the first injectable, biodegradable microparticle depot system introduced by Debiopharm into the European market for prostate cancer treatment. The active pharmaceutical ingredient (API), triptorelin, is a synthetic decapeptide agonist analog of luteinizing hormone releasing hormone (LHRH). Later in 1989, FDA approved PLGA microspheres (Lupron Depot®) as an alternative treatment for advanced prostate cancer, which contained synthetic nonapeptide (leuprolide acetate/leuprorelin) as one of the initial products for sustained peptide delivery. In recent ten years, implanted PLGA products Bydureon® and Signifor® LAR have been approved in 2012 and 2014 respectively for long-acting peptides delivery. Bydureon® is comprised of PLGA microparticles that encapsulate exenatide for antidiabetic usage and needs to be administered subcutaneously. Signifor® LAR is a pasireotide-loaded PLGA microparticle suspension for treating Cushing's disease and acromegaly. After re-suspension, immediate intramuscular injection in the gluteus is required.

|
Download:
|
| Fig. 2. Timelines of injectable/implantable PLGA-based products in market. The Decapeptyl® is the first clinically approved PLGA product in the market developed by Ipsen pharmaceutical. Effective component and the indication for each product are mentioned on branches. Peptide/protein products are highlighted with yellow background. | |
Inspired by the successful marketing of PLGA products, more PLGA-based implants have paved their way to clinics in recent years. According to ClinicalTrails.gov [27], more than thirty trials are completed or still ongoing with different clinical phases (Table 1). The purpose of these clinical trials is either to explore existing products on other indications or to investigate the potential of PLGA implants for sustained delivery of newly approved therapeutic agents. NCT04094298 is an ongoing phase III trial for assessing the overall safety and tolerability of TA-ER/FX006, which contains triamcinolone-loaded PLGA microsphere to treat rotator cuff disease. CEB-01 is a newly developed antineoplastic PLGA membrane loaded with SN-38 and could be implanted on resection surfaces infiltrated by tumor cells. During the phase I clinical trial that started in 2020, the safety and tolerability will be evaluated in patients with recurrent or locally advanced retroperitoneal soft tissue sarcoma after surgery (NCT04619056). Somatuline®, PLGA-based injectable microparticle, which has been approved by FDA to treat acromegaly in 2007, was introduced in a phase II clinical trial for unresectable and/or metastatic Merkel cell carcinoma treatment.
|
|
Table 1 Completed and undergoing clinical trials on injectable/implantable PLGA formulations in twenty years. |
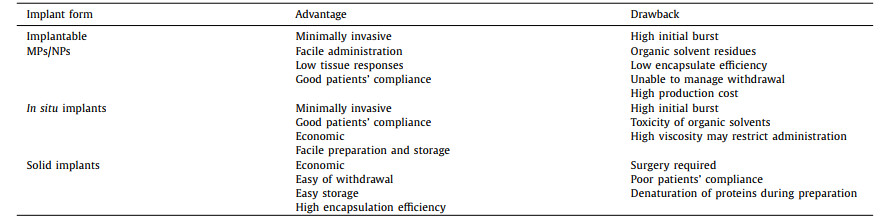
PLGA-based implants have various forms, such as implanted microspheres/nanoparticles, injectable in-situ hydrogels, and solid implants. And their characteristics, including advantages and drawbacks are summarized in Table 2. The main differences between these implants lie in preparation methods, production expenses, administration route, drug loading and patients' compliance. By far, various technologies have been developed to fabricate these implants.
|
|
Table 2 Characteristics of various PLGA-based implants. |
{kind=link}
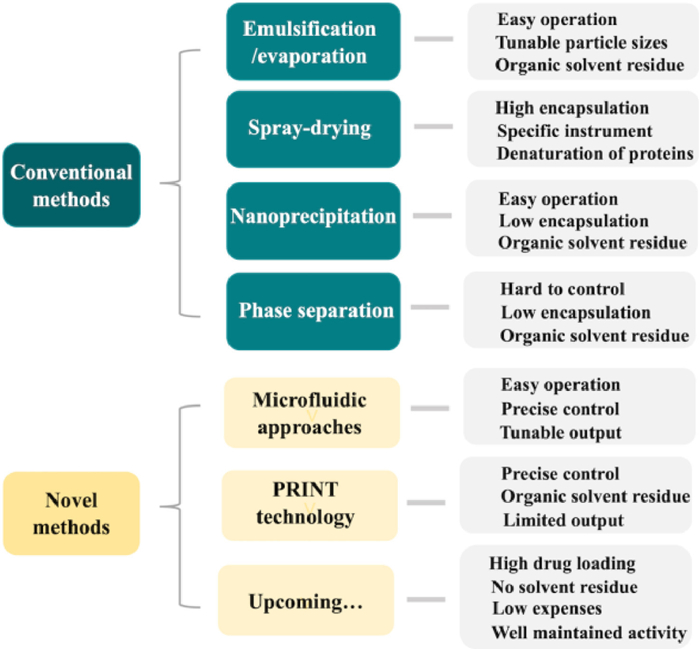
Polymer microparticles (MPs) are defined as particles of any shape with particle sizes ranging from 100 nm to 100 µm while nanoparticles (NPs) are particles smaller than 100 nm [16,28]. Owing to their capacity of suspending in aqueous solutions, implanted MPs/NPs can be injected without anesthesia, which largely improves patients' compliance [29]. According to the physicochemical feature of loaded peptides/proteins and PLGA copolymer, various techniques have been applied to prepare peptide/protein-loaded MPs/NPs. The conventional methods include emulsification-solvent evaporation, spray-drying, nanoprecipitation, and phase separation, while novel fabrication techniques, such as microfluidic approaches and PRINT technology, have been also developed in recent decades (Fig. 3). Among marketed products, Sandostatin® and Bydureon® are manufactured through coacervation, while Somatuline® Depot and Trelstar® are fabricated by spray drying technology. Besides, Signifor® LAR as well as Lupron Depot® involve the emulsification-solvent evaporation technology.

|
Download:
|
| Fig. 3. Fabrication technologies for PLGA MPs/NPs and the corresponding characteristics. | |
{kind=link}
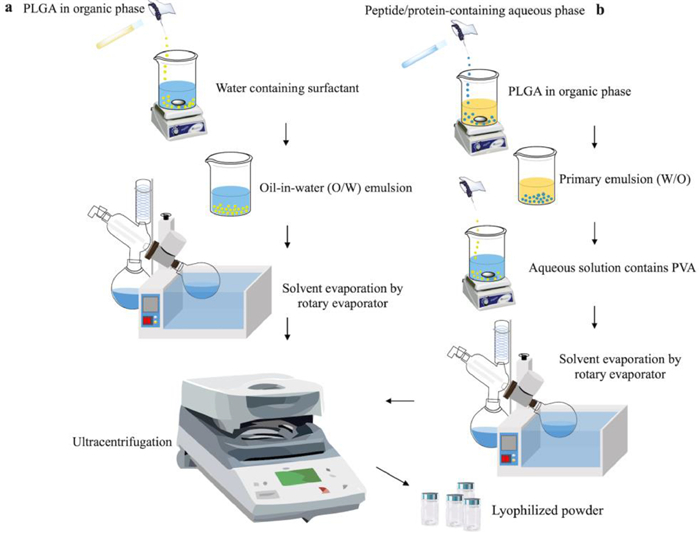
As the most common method to prepare peptide/protein-loaded MPs/NPs, emulsification-solvent evaporation methods can be categorized into single and double emulsion solvent evaporation methods (Fig. 4). Polyvinyl alcohol (PVA) is frequently used as the surfactant or stabilizer as it could be removed conveniently [30,31]. Oil-in-water (O/W) emulsification is a typical example of the single emulsion process. Briefly, the polymer is firstly dissolved in an organic solvent that is water-immiscible and volatile, such as dichloromethane (DCM). The drug is then added to the organic phase to produce a dispersion before adding PVA aqueous solution for emulsion at the appropriate temperature with stirring. Single emulsion solvent evaporation method is commonly utilized for encapsulating small molecular hydrophobic chemicals or hydrophobic peptides, while water-in-oil-in-water (W/O/W) methods are preferred to encapsulate water-soluble peptides/proteins. For instance, proteins are dissolved in deionized water and then added to the organic phase containing PLGA. The primary emulsion is added to the PVA aqueous solution to further emulsify at appropriate conditions. The organic solvent, either from single or double emulsion methods, could be evaporated or extracted. The prepared MPs/NPs are then separated by ultracentrifugation and freeze-dried. The size of final particles and encapsulation efficiency could be adjusted by altering emulsify condition, solvent and the stirring rate [32]. The structure of MPs could be tuned by adjusting protein concentration in the aqueous phase. In a study conducted by Zhang et al., high bovine serum albumin (BSA) content usually leads the formation of porous microspheres while low BSA content confer a hollow porous structure [33]. In addition to peptides and proteins, the double emulsion method has been extensively utilized for fabricating NPs for anticancer therapies. It has been evidenced that the hemin and lipoxidase co-loaded CaCO3-encapsulated PLGA nanoreactor (HLCaP NRs) could suppress the growth of residual tumors using tumor debris as the fuel [34]. A PLGA-based nanocarrier has been developed for transporting Na+ and Cl− to induce apoptosis via W/O/W double emulsion, which shows great potential to eliminating cancer cells [35].

|
Download:
|
| Fig. 4. The schematic illustration of the single emulsion solvent evaporation method (a), and double emulsion solvent evaporation (b). | |
{kind=link}
Unlike the emulsion method that requires precise and complicated control for high encapsulation efficiency and mass production, spray-drying is suitable for scalable industrial production with a few process parameters such as the orientation of jets, and material flow rates as well as the rate of solvent extraction [36]. Hydrophobic drugs are usually dissolved in organic solvent such as DCM, while hydrophilic proteins are dissolved in aqueous solvent before emulsified with polymer-containing organic solution. Then, the water-in-oil emulsion is subjected to a hot gas stream for solvent evaporation. The feed rate and the temperature during the solvent evaporation influence the morphology of obtained PLGA particles [37]. However, the high temperature often leads to the denaturation and aggregation of proteins which usually occurs at the water/oil interface. As a result, solid-in-oil dispersion has been applied as an alternative method during the spray drying due to the relatively high stability of dehydrated proteins in neat organic solvents and their thermostability [38]. Additionally, PEG could form complexes with proteins in pure DCM directly without introducing water, facilitating the production of low-initial-burst PLGA microparticles using spray drying [39].
4.1.3. NanoprecipitationNanoprecipitation is one of the most popular methods for NPs fabrication and has also been used for preparation of peptide/protein encapsulated PLGA NPs in past years. Commonly, PLGA polymers are dissolved in water-miscible organic solvent while the protein drug is added to the aqueous solution containing emulsifiers. The polymer solution and the drug are diffused to the aqueous solution dropwise under constant agitation. The organic solvent such as acetone could be easily removed by reduced pressure evaporation. The particle size of NPs obtained by nanoprecipitation is usually around 200 nm which is smaller than the counterparts produced via other approaches [40]. The most critical parameter for this methodology is the solvent miscibility, while solvent with good solubility in water often produces NPs with smaller size. The size and drug loading efficiency could be tuned either by adjusting the rate of polymer addition or the stirring speed [41]. Unfortunately, owing to the hydrophilicity nature of protein drugs, the encapsulation efficiency of this technique is always limited.
4.1.4. Phase separationPhase separation, also called coacervation, fabricates peptide/protein-loaded PLGA MPs by liquid–liquid phase separation. Briefly, PLGA polymers are dissolved entirely in the organic solvent, and the protein drugs in solid form are then added. Afterwards, silicon oil is added into the primary dispersion at a defined rate to decrease the solubility of PLGA, which induces the coacervation. The PLGA-rich phase encapsulates surrounded protein molecules to form microspheres, which finally solidify by transferring into a heptane medium to remove solvents. Rinsed by water, the collected MPs are sieved and dried to obtain the final product. For example, protein diphtheria tocoid was encapsulated by this technique [42]. It is noteworthy that encapsulation efficiency of peptides/proteins is subject to the compatibility between the PLGA and the coacervation agent [6].
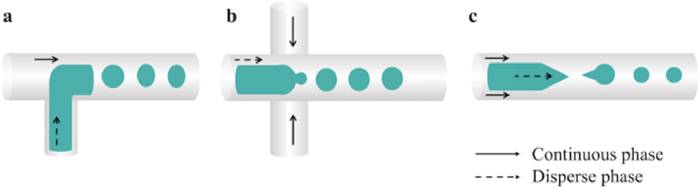
4.1.5. Microfluidic approachesThe emerging microfluidic technology is a versatile alternative to conventional methods for producing uniform PLGA MPs/NPs. Different materials could be used on microfluidic devices, such as glass, silicon, perfluoropolyether, polydimethylsiloxane (PDMS), and polymethyl methacrylate (PMMA) [43,44]. Small volumes of fluids ranging from microliters to picoliters are driven through microchannel networks with various shapes, including the T-junction channel, the co-flow channel, and the flow-focusing channel (Fig. 5). Immiscible continuous and dispersed phases are injected at independent entrances and then confined into isolated droplets or narrow streams. The size and chemical composition could be altered by controlling the flow rate and the formulation of constituent fluid separately. Particles are assembled at the intersection of channels due to the high shear force. The advantages of microfluidic method include high production efficiency, low cost and facile operation. It allows precise control of fabrication of peptide/protein-loaded MPs from small amount to large-scale [45]. Besides, the electrostatic forces could be incorporated into microfluidics to control the morphology of drug-loaded particles [46]. To achieve the demand of mass production, some improvements have been adapted for microfluidic methodology. For instance, chips containing multiple droplet generators and a single droplet generator with multiple layers of geometry structure are used to improve the production efficiency [47,48].

|
Download:
|
| Fig. 5. The schematic illustration of microfluidic devices for fabricating PLGA MPs/NPs via T-junction microfluidic device (a), co-flow microfluidic device (b), and flow-focusing microfluidic device (c). Arrows with real lines indicate continuous phases, and arrows with dashed lines represent disperse phases. | |
{kind=link}
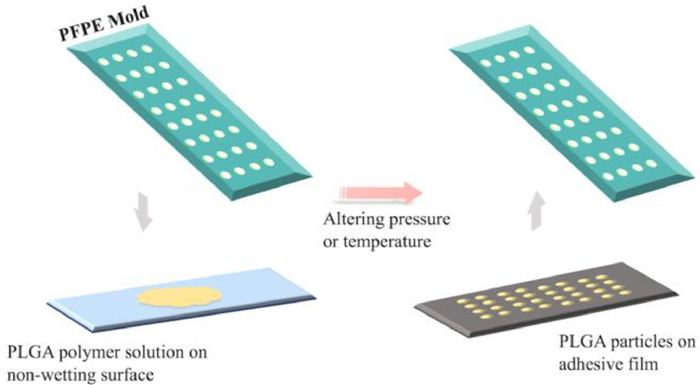
Particle replication in non-wetting templates (PRINT) was firstly introduced in 2005 and is a continuous top-down technology to produce monodisperse MPs/NPs with precise structure [49]. A non-wetting elastomeric mold containing wells or cavities of predefined shape and size is pressed against the cargo-containing polymer solution on another non-wetting surface. After solidification by altering pressure or temperature, particles with sizes ranging from 80 nm to 20 µm could be recovered with various structures using the adhesive film (Fig. 6). Generally, the PRINT technology utilizes perfluoropolyether (PFPE) material to make template mold due to its low toxicity and organic-solvent-tolerant capacity. Besides, the inherits minimal surface energy of PFPE facilitates the detachment of particles from the mold surface. It prevents the overflow on non-cavity regions, leading to the formation of well-isolated MPs/NPs. Similar to microfluidic approaches, PRINT technology could easily achieve the translation from laboratory to market as it is compatible with GMP requirements [50].

|
Download:
|
| Fig. 6. Particle replication in non-wetting templates (PRINT) technology. A non-wetting elastomeric mold containing predefined cavities is pressed against the cargo-containing polymer solution on another non-wetting surface. After the solidification by altering pressure or temperature, particles could be recovered by applying the adhesive film. | |
{kind=link}
During preparation of the PLGA-based MPs/NPs, the active protein ingredients are encapsulated by dissolving into the organic or aqueous solution, using ultrasonic devices or high shear homogenization. Moreover, the desired peptide/protein could be loaded to the blank particles by incubation overnight under defined conditions at 4 ℃. The protein-absorbed MPs/NPs could be further separated from free protein solution by ultra-centrifugation or gel filtration. Then the particle pellets are ready for washing and lyophilization prior to use.
4.1.7. CharacterizationTo characterize physical/chemical properties of fabricated protein-loaded particles, a variety of analytical tools are employed. Nuclear magnetic resonance (NMR) and differential scanning calorimeter (DSC) are applied to analyze the solid state of PLGA polymer as well as the ratio of lactic and glycolic acids and end groups. Optical microscopes help to obtain a quick overview of the size and shape of the PLGA particles, while scanning electron microscopy (SEM) could further determine the surface morphology and structure. Porosity analyzer can be utilized to determine the pore size and pore distribution on MPs/NPs. The conformational structure and the distribution of protein drugs loaded in the matrix could be detected by Raman spectroscopy. The high-performance liquid chromatography (HPLC) and ultra-performance liquid chromatography (UPLC) help to characterize and quantify the released/degraded effective components. To quantify loaded peptides or proteins, the UV–visible spectrophotometer, HPLC and other methods could be involved. And sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) can help to investigate the structural integrity of peptide/protein drugs [30].
4.2. PLGA-based in situ forming gelsIn-situ forming implant (ISFI) systems are described as polymer-based systems initially comprised of the liquid solution which can be solidified after being administered subcutaneously or intramuscularly [51]. Peptide/protein drug is dissolved or suspended in a concentrated solution of PLGA polymer and water-miscible biocompatible solvent. The active component could be entrapped within the polymer matrix during the precipitation when the organic solvent dissipates into the surrounding external aqueous body fluids. According to the mechanism of solidification, ISFI systems have been classified in cross-linking systems [52], in-situ solidifying organogel systems [53] and the widespread in-situ phase separation systems [54]. Thanks to its facile manufacture, enhanced patient compliance and economic benefits, a few PLGA-based ISFI products for sustained peptide/protein release have been introduced to the market, such as Eligard®, Lupron DepotTM, and Sandostatin LAR®. Eligard® is designed to deliver leuprolide acetate subcutaneously for prostate cancer treatment [55]. With the same component, Lupron DepotTM works as an intramuscular ISFI product offering variable dosing regimens for treating advanced prostate cancer. Lupron Depot-PedTM is another commercially available product of Lupron DepotTM for the treatment of central precocious puberty in 1–3 months infants [56]. Sandostatin LAR®, loaded with octreotide, is prescribed as a monthly intramuscular ISFI product to control the growth hormone levels and the gastrointestinal hormone secretions [57].
Solvents employed in AtriGel® range from hydrophilic solvents (i.e., dimethyl sulfoxide, N-methyl-2-pyrrolidone (NMP), tetra glycol, and glycol furol) to hydrophobic solvents (i.e., ethyl acetate, triacetin, and benzyl benzoate). The lyophilized drug-containing powder is commonly filled in one syringe and necessitate mix with the polymeric solution (PLGA/NMP) present in the other syringe to form a homogeneous combination before administration [58-60].
Compared with MPs/NPs, PLGA-based ISFI systems have distinct properties which need to be well characterized. Generally, the clarity test apparatus could be utilized to determine the presence of undesirable components in the ISFI formulation, while viscosity and rheology can be assessed using Brookfield rheometer or other type of viscometers as ISFI systems with suitable viscosity should guarantee no difficulties during its administration to patients. The syringeability is a key mechanical parameter to be considered during the preparation of ISFI system and could be evaluated via the texture profile analyzer. Moreover, the gel strength is an important indication of the system viscosity under physiological conditions to describe the stiffness of gel networks, which could be measured by Rheometer. Ideal gel strength contributes to easier control of the drug release owing to the none leakage from the injection site [61,62]. The hardness is determined as the maximum compression force to deform the sample [63]. Low hardness express good spreadability and easy administration of the system, while ISFI formulations with high hardness, adhesiveness and cohesiveness display a slow and controlled drug release.
4.3. PLGA-based solid implantAs an alternative to the solvent-based production methods, PLGA materials are moldable via thermal processing in solid and melt state. Not only for small-scale fabrication, this solid-based formulation technology is also suitable for industrial production owing to its high throughput and continuous operation without compromising the encapsulation efficiency. Generally, proteins are loaded into PLGA particles before or during the sintering processing prior to assembling together into implantable devices. The melting temperature (Tm) and glass transition temperature (Tg) of PLGA copolymer depend on the percentage of glycolic acid, with the range of 80–160 ℃ and 33–60 ℃, respectively [64]. The 50:50 PLGA which exhibits the lowest Tm and Tg has been widely applied for thermal processing [12]. Nevertheless, elevated temperature and pressure may lead to the denaturation or aggregation of peptide/protein components. Viscosity modifiers such as PEG or sugars could help to decrease the required temperature during the molding. For instance, a novel PLGA/PEG scaffold loaded with BMP-2 allows a reduced production Tg around 37 ℃. It provides a promising surgical tool for bone tissue engineering of craniomaxillofacial repair [65].
Hot-melt extrusion (HME) has been predominately used in peptide/protein encapsulation by melting the mixture of API and solid PLGA with conveying screw until it is forced through a die and solidified [66]. To avoid the detrimental effects brought by high shear force and high temperature during the HME process, solid proteins have better performance on maintaining correct conformation than proteins in aqueous state. When water is removed, Tg shifts from the limited 30 ℃ to 130–185 ℃, which is beneficial for fabricating solid state implants due to the restricted molecular motion [67]. There is also risk of thermal-induced acylation or thioester formation between PLGA and the surface residues of protein during melt processing [68]. To achieve the stabilization of protein structure, additives such as PEG or sugar align along the protein/polymer interface to shield the protein from the hydrophobic polymer melt [69].
Ghalanbor et al. have proved that lysozyme, a widely utilized model protein, could be completely released from different formulations while the release rate was controlled by the protein loading and additives. And DSC results indicated that the protein conformation conserved at 105 ℃ during the extrusion, which is lower than the denaturation temperature 204 ℃. The spectra of 25% lysozyme loaded into implant was comparable to the raw lysozyme power. As a consequence, there was no evidence for any denaturation or aggregation of protein during the molding process [70].
PLGA-based implants have also been explored to delivery vaccines as well as therapeutic proteins (e.g., antibodies) recently. For instance, HPV vaccines can be encapsulated in PLGA matrix using a bench top melt-extrusion equipment. It was reported that the immunogenicity of protein conjugate HPV-Qβ was well-maintained during the melt extrusion process. The titer of IgG generated by single-dose HPV-Qβ/PLGA implant was comparable to traditional soluble vaccines [71]. Efforts have been devoted to combining HME with 3D printing method to produce protein-loaded devices for achieving tissue engineering and drug delivery simultaneously [72-74]. Recently, Carlier et al. developed a monoclonal antibodies (mAbs)-loaded 3D-printed PLGA implant. The mixture of PEG, PLGA and mAb power was extruded into printable filaments to feed 3D printer. The stability of mAb against thermal degradation has been improved by using a lower amount of l-leucine in the formulation. Additionally, d-(+)-trehalose dihydrate and l-leucine help the proteins maintained in small molecule and high released feasibly. Thus, despite the relative high temperature during the extrusion and printing process (90 and 105 ℃), over 70% of binding capacity of mAb was maintained up to 24 h after dissolution [72].
5. Encapsulation efficiency, drug release and influencing factors 5.1. Encapsulation efficiency (EE%) of PLGA-based implantsEE% is one of important parameters for long-acting delivery systems. Commonly, the EE% of peptides/proteins in implants is influenced by the fabrication technologies, constituents, and properties of PLGA polymers. For example, the EE% in PLGA NPs/MPs prepared by emulsion methods is always limited, and predominately affected by stirring rates and solvents [5]. However, replacing the external water phase in W/O/W emulsification with an oil phase immiscible with the polymer organic phase can prevent the protein diffusion and further facilitate encapsulation [75]. Compared with emulsion methods, the spray-drying and microfluidic approaches could enable a relative high EE%, while the nanoprecipitation always confers a low EE%. To achieve a high encapsulation, an adaption has been reported, with a first protein nanoprecipitation followed by a second precipitation of PLGA [76].
During the emulsification, the immiscibility of the organic solvent could affect the initial extraction of PLGA and thus EE%. For instance, MPs prepared with DCM display a higher EE% than counterparts made with ethyl acetate [77]. The removal of solvent also affects the encapsulation efficiency. A rapid process favors a high encapsulation. According to the Cho's research, EE% varied from 10% to 75%, which ethyl acetate quenching time was tuned from 45 min to 5 min [78]. Appropriate excipients such as stabilizers could also contribute to the high encapsulation. With the addition of dextran sulfate sodium (DSS) and diethyl phthalate (DEP), the EE% of octreotide acetate in PLGA could be enhanced to 90% [79]. In contrast, surfactants such as poloxamer 188 and 331 may compromise the encapsulation of proteins [80]. In addition to the additive type, additive concentration impact drug encapsulation as well. It has been reported that PVA concentration at 1% (w/v) provides the highest EE% compared with ones at 0.5% and 2% (w/v) [81].
The fundamental properties of PLGA polymers also play an important role in encapsulation efficiency. It has been observed that NPs formulated with PLGA with the molecule weight of 80 kDa have the highest EE% of 98% among others [82]. This is mainly ascribed to the fact that high intrinsic viscosity of PLGA polymer could enhance the encapsulation process by decreasing the diffusion of peptide/protein molecules [9]. Besides, the presence of uncapped carboxylic end groups of PLGA could enhance EE% by the ionic interaction between negatively charged carboxylic acids and positively charged proteins [82]. And conjugating PLGA with PEG usually results in the reduction of EE%, which is mainly due to the steric hindrance on protein–polymer interaction [83].
5.2. Drug release of PLGA-based MPs/NPs 5.2.1. Polymer-related factorsIt has been well acquainted that encapsulated peptides and proteins can be released from PLGA matrix by diffusion and erosion [84]. Any factors that influence the degradation of PLGA can affect drug release. Therein, the primary characteristics needed to be considered are the lactide/glycolide (L/G) ratio and molecular weight. In detail, they could impact the hydrophilicity, hydration rate and Tg of the PLGA polymer. Polylactide displays slower degradation than polyglycolide, owing to its less hydrophilicity. Therefore, high lactide/glycolide (L/G) ratio confers a slow degradation of PLGA, and thus a long drug release duration. Besides, PLGA polymers with larger molecular weights demonstrate a longer drug release duration than counterparts with smaller molecular weights [85,86]. The melting point and the degree of crystallinity are both directly related to the molecular weight [10]. Another factor that could influence degradation is the end group of PLGA copolymer. For instance, the presence of free carboxylic acid moieties will impact the water uptake and accelerate the rates of hydrolysis and erosion, which shortens drug release period [87]. Therefore, the drug release rate and duration in implants are commonly impacted by a variety of processes including the surface diffusion, surface erosion, bulk diffusion, bulk erosion and drug dissolution. Generally, the drug release of peptides and proteins often follows a biphasic curve which is consisted of an initial burst followed by a progressive drug release phase.
The release of peptides and proteins from the polymer matrix can be influenced by the interaction between polymers and drugs, as well. It has been reported that positively charged peptides loaded in a negatively charged polymer matrix show slow-release profiles over a long period [88]. Besides, peptides and proteins can be acylated during the degradation process, which leads to the formation of peptide-PLGA adducts by involving the nucleophilic attack process between peptide residues and electrophilic carbonyl groups [89]. Newly formed covalent bonds between peptides and PLGA can sustain the release of peptides and proteins, as well [90]. Strategies have been applied to minimize the compromised drug release associated with acylation. Additives such as sodium do-decyl sulfate (SDS), dextran sulfate A (DSA), and dextran sulfate B (DSB) could mask the reactive nucleophile amine of octreotide, forming reversible hydrophobic ion-pairing (HIP) complex [91]. While additional water-soluble dicationic salts, such as Mn2+ and Zn2+, could inhibit acylation by shielding the binding sites on PLGA and peptide/protein drugs [92]. Besides, PEGylation of peptides and proteins has been also reported to reduce the acylation [93].
5.2.2. Protein-related factorsThe denaturation and aggregation of peptides and proteins are commonly thought to be the important factors that confer a sustained and even incomplete release [94]. It has been reported that the interfacial force between the water and organic phase induces the denaturation and aggregation of peptides and proteins [95]. Proteins containing intermolecular antiparallel beta-sheets are vulnerable to the hydrophobic absorbent from the oil/water interface [96]. To combat this, various strategies have been developed. For instance, Hong et al. have introduced a hydrophilic oil phase to reduce the hydrophobicity as a novel W/O/Oh/W emulsion to formulate long acting injectable PLGA-based microspheres [97]. Stabilizers such as surfactants could also be applied to protect proteins from denaturation [98]. Besides, a solid-in-oil-in-water (S/O/W) emulsion has been reported, formulating peptide/protein as 1–10 µm freeze-dried bulk powder to prevent the conformational change [99,100].
5.2.3. System-related factorsOther factors, such as size and morphology, can contribute to the degradation and drug release duration [101]. According to Siepmann et al.'s results, the diffusion pathways' length increases when the average diameter increases from 7.2 µm to 53 µm, leading to a decrease in the drug release rate [102].
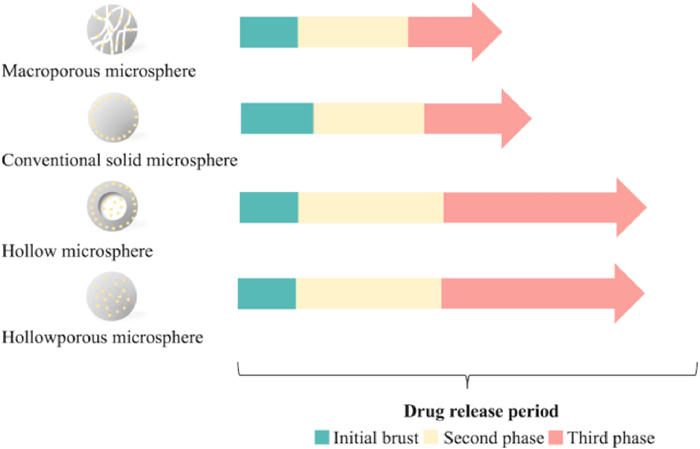
The morphology of implanted MPs/NPs may impact the diffusion pathway, and thus affect stages during the drug release period. Ma et al. prepared microspheres with various morphologies and loaded BSA as the model protein (Fig. 7). And their results revealed that the macroporous microspheres and conventional solid microspheres have the highest initial burst and rapid release rate, due to large pores and surface absorption, respectively [103]. For hollow microspheres, BSA was released with an initial burst from the shell followed by a much slower second burst for 60 h, and 85% of drug was released in the third stage. For hollow-porous microspheres, the initial burst was controlled within 20% and most BSA followed a slower linear release profile from the interior by diffusion due to the concentration gradient. It has been evidenced that the severe initial burst induced by surface-enrichment particles could be tuned by different preparation methods [104].

|
Download:
|
| Fig. 7. The illustration of microspheres with different morphologies and the drug release rates. Light yellow spheres in the figure represent peptide/protein drugs loaded into PLGA microspheres. The lengths of arrows indicate the duration of various drug release stages. | |
{kind=link}
Extreme environmental factors also threaten the integrity of drug-loading PLGA particles, and thus affect drug release. In general, microspheres are stable in about 12 months below glass transition temperature. It has been proposed that humid conditions and elevated temperature could influence the chain mobility and lead to a different glass transition temperature, therefore introduce to undesirable drug release behavior. In contrast, low storage temperature brings negligible physical change with time [105,106].
5.3. Drug release of PLGA-based in situ gelsDistinct from PLGA MPs/NPS, the drug release process of the ISFI systems typically involves three stages, that is, an initial burst during the solidification, a second stage via diffusion, and subsequent drug release via erosion of the polymeric matrix [107].
The solidification relies on the kinetics of the exchange between the solvent and non-solvent (i.e., body fluids). This phase inversion dynamics and the initial burst drug release can be controlled by altering the composition of the ISFI system. Liu et al. pointed out that the hydrophilicity of the polymer is negatively correlated to accelerate phase inversion and drug release [108]. In contrast, highly hydrophobic PLA copolymer without glycolic acid could form a highly porous structure and undergo faster precipitation. And the solubility, diffusivity, dissociation constant, and molecular weight of studied drug can influence the initial burst release, as well [109].
The subsequent diffusion stage mainly relies on the physio-chemical properties of active component, the polymer and the morphology of the matrix. Polymers with lower molecular weight and more flexible polymeric chain allow higher diffusivity of peptides and proteins in the system. Studies conducted by Mittal et al. revealed that the diffusion of drug through PLGA with low molecular weight followed zero-order kinetics, while drug diffusion through PLGA with higher molecular weight followed a Higuchi model with constant diffusion coefficients [110]. In another study, Ibrahim et al. pointed out that the increasing PLGA concentration and viscosity could reduce the drug release in the diffusion stage [62].
5.4. Drug release of PLGA solid implantsSimilar to microparticle systems, the release profiles and the stability of proteins released from solid PLGA implants depend on protein properties, protein loading, protein dispersion as well as additives [111]. Bulk implants have lower specific surface area than PLGA MPs/NPs. Therefore, the solid bulk implant requires extensive water uptake for degradation and elicits pore formation allowing diffusion of peptides and proteins. Increased protein loading could confer rapid release because abundant proteins are localized at the surface layer and released during the early hydration and degradation phases. Noteworthily, incorporation with additives such as hydrophilic porogens, can accelerate the release of peptides and proteins. The basic metal salts could neutralize the acid environment during the PLGA degradation, and attenuate the aggregation of proteins, allowing the fast diffusion of monomeric proteins through the implant [70]. The aggregation can also be adjusted by controlling initial particle size of solid protein before melting with PLGA [69]. Besides, modified protein with amphiphilic or hydrophobic moieties exhibits higher release rate [112].
6. Challenges and future perspectivesWith the rapid advancement of biotechnology, the role of peptides and proteins in diagnostic, therapeutic and preventive medicine is increasing. However, most of commercial peptides and proteins are administrated by repeatedly injection, which compromises patients' compliance. Concert efforts have been devoted to develop novel drug delivery systems for improving bioavailability, stability as well as targeting efficiency of peptides and proteins, while PLGA implants have gained more and more interests in recent years. Thanks to their excellent biodegradability and biocompatibility, decades of products have been approved for clinical use. In spite of various technologies developed for fabrication of PLGA based implants, the obtained PLGA implants are far from satisfaction.
Firstly, the drug loading efficiency of peptides and proteins in PLGA implants is not adequate. For instance, the nanoprecipitation technology and phase separation are commonly thought to be unsuitable for mass production owing to their low encapsulation efficiency. Although the drug loading efficiency could be tuned by adjusting parameters, such as the rate of polymer addition, the amount of dissolved/solid protein that can be loaded into the polymer matrix is limited as both of the methods rely on random incorporation of surrounding proteins [41,113]. Secondly, safety issue remains when organic solvents are involved during the preparation process. This not only refers to the safety for manufacture, but also the toxicity of residual solvents. W/O/W emulsification-solvent evaporation are most commonly utilized for fabricating peptide/protein-loaded MPs/NPs owing to the relative high encapsulation efficiency, whereas the organic solvents such as DCM are hard to remove completely. Other technologies, such as phase separation and microfluidic approaches, also suffer from such safety concern [94].
The instability of proteins becomes another issue associated with the safety and the cost. Denatured proteins usually lose the therapeutic efficiency or even trigger dangerous immune response. During the manufacture, the interfacial force between water and organic phases may lead to the undesired denaturation. Spray drying and hot melt extrusion could be alternatives for the organic solvent-based fabrications. Nevertheless, the inevitable elevated temperature is also harmful to the protein conformation. In addition, extreme environmental factors during the storage and degradation threaten the stability of peptide/protein-loaded PLGA implants, as well. Humid conditions and acidic environment would influence the chain mobility and therefore lead to the protein aggregation and loss of activity [105]. After implanting in vivo, inner micro environmental pH may change along with the degradation of polymers, and would confer a degradation and aggregation of protein drugs.
Another challenge is the initial burst release, which is not only common in MPs/NPs, but also in situ gels and solid implants. It has become one of the most intractable safety issues in long-acting delivery systems for years. Encapsulated peptides/proteins are released rapidly within a short period following the administration, which would compress the therapeutic duration, or at worse threaten lives [9]. This is mainly ascribed to the large pores within implants and abundant surface absorption of peptides and proteins. Furthermore, the increased protein loading could also lead to a rapid release as more proteins are localized to the surface layer [102]. For in situ hydrogels, the initial burst is also associated with phase inversion dynamics and basic characteristics of the protein drugs [108,109].
Finally, we believe that the quality of PLGA implants can be improved by overcoming these challenges. For instance, developing novel biocompatible additives may help to enhance the drug loading of peptides/proteins. Noteworthily, merely pursuing high drug loading may be inappropriate, as high drug loading are always associated with staggering burst release. Therefore, pursuing a balance between the drug loading and burst release may be a feasible optimizing strategy for PLGA implants currently. As organic solvents are inevitable during the fabrication of PLGA implants, exploiting green and safe solvents, such as ionic liquids can also help to improve the quality of PLGA implants [114-116]. As for industrial manufacture, manufacture methods with tunable output, such as microfluidics hold the potential to facilitate the economical production. By further optimize these methodologies, the quality of PLGA implants can be further improved, and the total expenses including production and storage could be significantly reduced.
In addition, developing more accurate methodologies for characterizing PLGA implants in vitro and in vivo, including degradation, drug release, etc., can also help to optimize PLGA implants, and is believed to hold the key to overcome these challenges. By dissecting the in vitro and in vivo processes of PLGA implants, we can further grasp the important parameters that influence the efficiency of peptides and proteins, moreover, find the way to optimize the formulation or fabrication technologies of PLGA implants. For example, developing biomimetic gel-based systems can help to accurately investigate drug release and predict long-term performance of implants in vitro [117,118]. To explore the in vivo process of PLGA implants, radio labeling and fluorescence labeling may be feasible monitoring strategies, while labeling with absolute aggregation-caused quenching probes can provide more accurate results owing to their excellent discrimination from signals of free probes [119-125]. Finally, we anticipate that PLGA implants can be an excellent drug delivery system for vaccines, owing to their long-term drug release. Confronted with the pandemics and potential recurrent infection of influenza as well as COVID-19, we believe that PLGA implants are capable of arming us with high titers of antibodies against these viruses in the long period.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors would like to acknowledge the financial support from National Natural Science Foundation of China (Nos. 82104082, 81973247 and 82030107), Shanghai Municipal Commission of Science and Technology (Nos. 19XD1400300 and 21430760800).
| [1] |
M.H. Asfour, Drug Deliv. Transl. Res. 11 (2021) 1-23. DOI:10.1007/s13346-020-00746-z |
| [2] |
B.J. Bruno, G.D. Miller, C.S. Lim, Ther. Deliv. 4 (2013) 1443-1467. DOI:10.4155/tde.13.104 |
| [3] |
D. Ibraheem, A. Elaissari, H. Fessi, Int. J. Pharm. 477 (2014) 578-589. DOI:10.1016/j.ijpharm.2014.10.059 |
| [4] |
R.F. Pagels, R.K. Prud'homme, J. Control. Release 219 (2015) 519-535. DOI:10.1016/j.jconrel.2015.09.001 |
| [5] |
H.K. Makadia, S.J. Siegel, Polymers 3 (2011) 1377-1397. DOI:10.3390/polym3031377 |
| [6] |
A. Butreddy, R.P. Gaddam, N. Kommineni, N. Dudhipala, C. Voshavar, Int. J. Mol. Sci. 22 (2021) 8884. DOI:10.3390/ijms22168884 |
| [7] |
X. Sun, C. Xu, G. Wu, Q. Ye, C. Wang, Polymers 9 (2017) 189. DOI:10.3390/polym9060189 |
| [8] |
K. Park, S. Skidmore, J. Hadar, et al., J. Control. Release 304 (2019) 125-134. DOI:10.1016/j.jconrel.2019.05.003 |
| [9] |
S.D. Allison, Expert Opin. Drug. Deliv. 5 (2008) 615-628. DOI:10.1517/17425247.5.6.615 |
| [10] |
X.S. Wu, N. Wang, J. Biomater. Sci. Poly. Ed. 12 (2001) 21-34. DOI:10.1163/156856201744425 |
| [11] |
M.A. Elsawy, K.H. Kim, J.W. Park, A. Deep, Renew. Sustain. Energy Rev. 79 (2017) 1346-1352. DOI:10.1016/j.rser.2017.05.143 |
| [12] |
M.L. Houchin, E.M. Topp, J. Appl. Polym. Sci. 114 (2009) 2848-2854. DOI:10.1002/app.30813 |
| [13] |
Y.P. Li, Y.Y. Pei, X.Y. Zhang, et al., J. Control. Release 71 (2001) 203-211. DOI:10.1016/S0168-3659(01)00218-8 |
| [14] |
B. Jeong, Y.H. Bae, S.W. Kim, J. Biomed. Mater. Res. 50 (2000) 171-177. DOI:10.1002/(SICI)1097-4636(200005)50:2<171::AID-JBM11>3.0.CO;2-F |
| [15] |
Y. Zhang, M. GarcíaGabilondo, A. Rosell, A. Roig, Pharmaceutics 12 (2019) 16. |
| [16] |
B. Semete, L. Booysen, Y. Lemmer, et al., Nanomedicine 6 (2010) 662-671. DOI:10.1016/j.nano.2010.02.002 |
| [17] |
Y. Zhou, A. Fang, F. Wang, et al., Chin. Chem. Lett. 31 (2020) 494-500. DOI:10.1016/j.cclet.2019.04.048 |
| [18] |
C. Xu, Y. Kang, X. Dong, D. Jiang, M. Qi, Chin. Chem. Lett. 34 (2023) 107528. DOI:10.1016/j.cclet.2022.05.042 |
| [19] |
X. Zhen, L. Li, L. Jia, et al., Chin. Chem. Lett. 34 (2023) 107680. DOI:10.1016/j.cclet.2022.07.023 |
| [20] |
L. Tang, M. Xie, J. Li, et al., Chin. Chem. Lett. 34 (2023) 107801. DOI:10.1016/j.cclet.2022.107801 |
| [21] |
C. Xu, Y. Kang, S. Guan, et al., Chin. Chem. Lett. 34 (2023) 107825. DOI:10.1016/j.cclet.2022.107825 |
| [22] |
J. Yuan, M. Guo, S. Zhao, et al., Chin. Chem. Lett. 34 (2023) 107943. DOI:10.1016/j.cclet.2022.107943 |
| [23] |
H. Liu, Z. Miao, Z. Zha, Chin. Chem. Lett. 33 (2022) 1673-1680. DOI:10.1016/j.cclet.2021.10.057 |
| [24] |
Y. Wang, B. Qin, G. Xia, S.H. Choi, AAPS J. 23 (2021) 92. DOI:10.1208/s12248-021-00611-y |
| [25] |
C. Zhang, L. Yang, F. Wan, et al., Int. J. Pharm. 585 (2020) 119441. DOI:10.1016/j.ijpharm.2020.119441 |
| [26] |
A. Jain, K.R. Kunduru, A. Basu, et al., Adv. Drug. Deliv. Rev. 107 (2016) 213-227. DOI:10.1016/j.addr.2016.07.002 |
| [27] |
U.S. National Library of Medicine, Available online: ClinicalTrails. gov (accessed on 8th Oct. 2022).
|
| [28] |
S. Slomkowski, J.V. Alemán, R.G. Gilbert, et al., Pure Appl. Chem. 83 (2011) 2229-2259. DOI:10.1351/PAC-REC-10-06-03 |
| [29] |
R. Jain, N.H. Shah, A.W. Malick, C.T. Rhodes, Drug Dev. Ind. Pharm. 24 (1998) 703-727. DOI:10.3109/03639049809082719 |
| [30] |
R.C. Mundargi, V.R. Babu, V. Rangaswamy, P. Patel, T.M. Aminabhavi, J. Control. Release 125 (2008) 193-209. DOI:10.1016/j.jconrel.2007.09.013 |
| [31] |
A. Kumari, S.K. Yadav, S.C. Yadav, Colloids Surf. B: Biointerfaces 75 (2010) 1-18. DOI:10.1016/j.colsurfb.2009.09.001 |
| [32] |
I.D. Rosca, F. Watari, M. Uo, J. Control. Release 99 (2004) 271-280. DOI:10.1016/j.jconrel.2004.07.007 |
| [33] |
G.H. Zhang, R.X. Hou, D.X. Zhan, et al., Chin. Chem. Lett. 24 (2013) 710-714. DOI:10.1016/j.cclet.2013.05.011 |
| [34] |
Z.Y. Zhu, Z. Dong, et al., Nanomicro Lett. 13 (2020) 29. |
| [35] |
C. Ma, Y. Zhang, Z. Jiao, et al., Chin. Chem. Lett. 31 (2020) 1635-1639. DOI:10.1016/j.cclet.2019.09.010 |
| [36] |
C. Berkland, E. Pollauf, D.W. Pack, K. Kim, J. Control. Release 96 (2004) 101-111. DOI:10.1016/j.jconrel.2004.01.018 |
| [37] |
N.Q. Shi, J. Zhou, J. Walker, et al., J. Control. Release 321 (2020) 756-772. DOI:10.1016/j.jconrel.2020.01.023 |
| [38] |
K.G. Carrasquillo, A.M. Stanley, J.C. AponteCarro, et al., J. Control. Release 76 (2001) 199-208. DOI:10.1016/S0168-3659(01)00430-8 |
| [39] |
H. Mok, T.G. Park, Eur. J. Pharm. Biopharm. 70 (2008) 137-144. DOI:10.1016/j.ejpb.2008.04.006 |
| [40] |
C. Vauthier, K. Bouchemal, Pharm. Res. 26 (2009) 1025-1058. DOI:10.1007/s11095-008-9800-3 |
| [41] |
C. Duclairoir, E. Nakache, H. Marchais, A.M. Orecchioni, Colloid Polym. Sci. 276 (1998) 321-327. DOI:10.1007/s003960050246 |
| [42] |
P. Johansen, L. Moon, H. Tamber, et al., Vaccine 18 (1999) 209-215. DOI:10.1016/S0264-410X(99)00191-7 |
| [43] |
R. Karnik, F. Gu, P. Basto, et al., Nano Lett. 8 (2008) 2906-2912. DOI:10.1021/nl801736q |
| [44] |
J.H. Xu, H. Zhao, W.J. Lan, G.S. Luo, Adv. Healthc. Mater. 1 (2012) 106-111. DOI:10.1002/adhm.201100014 |
| [45] |
G. Luo, L. Du, Y. Wang, Y. Lu, J. Xu, Particuology 9 (2011) 545-558. DOI:10.1016/j.partic.2011.06.004 |
| [46] |
I.G. Loscertales, A. Barrero, I. Guerrero, et al., Science 295 (2002) 1695-1698. DOI:10.1126/science.1067595 |
| [47] |
S. Nawar, J.K. Stolaroff, C. Ye, et al., Lab Chip 20 (2020) 147-154. DOI:10.1039/C9LC00966C |
| [48] |
H.Y. Peng, W. Wang, R. Xie, et al., Particuology 48 (2020) 74-87. DOI:10.1016/j.partic.2018.10.003 |
| [49] |
J.P. Rolland, B.W. Maynor, L.E. Euliss, et al., J. Am. Chem. Soc. 127 (2005) 10096-10100. DOI:10.1021/ja051977c |
| [50] |
J. Xu, D.H.C. Wong, J.D. Byrne, et al., Angew. Chem. Int. Ed. 52 (2013) 6580-6589. DOI:10.1002/anie.201209145 |
| [51] |
R.B. Patel, L. Solorio, H. Wu, T. Krupka, A.A. Exner, J. Control. Release 147 (2010) 350-358. DOI:10.1016/j.jconrel.2010.08.020 |
| [52] |
X. Li, Y. Wang, A. Li, et al., Molecules 24 (2019) 4211. DOI:10.3390/molecules24234211 |
| [53] |
A. Vintiloiu, M. Lafleur, G. Bastiat, J.C. Leroux, Pharm. Res. 25 (2008) 845-852. DOI:10.1007/s11095-007-9384-3 |
| [54] |
D.N. Kapoor, O.P. Katare, S. Dhawan, Int. J. Pharm. 426 (2012) 132-143. DOI:10.1016/j.ijpharm.2012.01.005 |
| [55] |
S.P. Schwendeman, R.B. Shah, B.A. Bailey, A.S. Schwendeman, J. Control. Release 190 (2014) 240-253. DOI:10.1016/j.jconrel.2014.05.057 |
| [56] |
R.R.S. Thakur, H.L. McMillan, D.S. Jones, J. Control. Release 176 (2014) 8-23. DOI:10.1016/j.jconrel.2013.12.020 |
| [57] |
J. Hadar, S. Skidmore, J. Garner, et al., J. Control. Release 304 (2019) 75-89. DOI:10.1016/j.jconrel.2019.04.039 |
| [58] |
M.J. Rathbone, J. Hadgraft, Modified Release Drug Delivery Technology, 1st ed, CRC Press, Boca Raton, 2002.
|
| [59] |
V. Halpern, R.M. Stalter, D.H. Owen, et al., Contraception 92 (2015) 3-9. DOI:10.1016/j.contraception.2015.02.014 |
| [60] |
C. Bode, H. Kranz, A. Kruszka, F. Siepmann, J. Siepmann, J. Drug Deliv. Sci. Technol. 53 (2019) 101180. DOI:10.1016/j.jddst.2019.101180 |
| [61] |
N. Kanwar, V.R. Sinha, Crit. Rev. Ther. Drug. Carrier Syst. 36 (2019) 93-136. DOI:10.1615/CritRevTherDrugCarrierSyst.2018025013 |
| [62] |
T.M. Ibrahim, N.A. ElMegrab, H.M. ElNahas, Pharm. Dev. Technol. 26 (2021) 709-728. DOI:10.1080/10837450.2021.1944207 |
| [63] |
V.G.R. Patlolla, W.P. Holbrook, S. Gizurarson, T. Kristmundsdottir, Gels 5 (2019) 47. DOI:10.3390/gels5040047 |
| [64] |
P. Lan, Y. Zhang, Q. Gao, H. Shao, X. Hu, J. Appl. Polym. Sci. 92 (2004) 2163-2168. DOI:10.1002/app.20197 |
| [65] |
C.V. Rahman, D. BenDavid, A. Dhillon, et al., J. Tissue Eng. Regen. Med. 8 (2014) 59-66. DOI:10.1002/term.1497 |
| [66] |
P.W. Lee, J.K. Pokorski, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 10 (2018) e1516. DOI:10.1002/wnan.1516 |
| [67] |
L.H.G. van Donkelaar, J.T. Martinez, H. Frijters, et al., Food Res. Int. 72 (2015) 241-246. DOI:10.1016/j.foodres.2015.03.042 |
| [68] |
Z. Ghalanbor, M. Körber, R. Bodmeier, Int. J. Pharm. 438 (2012) 302-306. DOI:10.1016/j.ijpharm.2012.09.015 |
| [69] |
K. Rajagopal, J. Wood, B. Tran, T.W. Patapoff, T. Nivaggioli, J. Pharm. Sci. 102 (2013) 2655-2666. DOI:10.1002/jps.23634 |
| [70] |
Z. Ghalanbor, M. Körber, R. Bodmeier, Pharm. Res. 27 (2010) 371-379. DOI:10.1007/s11095-009-0033-x |
| [71] |
S. Shao, O.A. OrtegaRivera, S. Ray, J.K. Pokorski, N.F. Steinmetz, Vaccines 9 (2021) 66. DOI:10.3390/vaccines9010066 |
| [72] |
E. Carlier, S. Marquette, C. Peerboom, K. Amighi, J. Goole, Int. J. Pharm. 597 (2021) 120337. DOI:10.1016/j.ijpharm.2021.120337 |
| [73] |
A. Goyanes, A.B.M. Buanz, G.B. Hatton, S. Gaisford, A.W. Basit, Eur. J. Pharm. Biopharm. 89 (2015) 157-162. DOI:10.1016/j.ejpb.2014.12.003 |
| [74] |
J. Holländer, N. Genina, H. Jukarainen, et al., J. Pharm. Sci. 105 (2016) 2665-2676. DOI:10.1016/j.xphs.2015.12.012 |
| [75] |
T.K. Giri, C. Choudhary, Ajazuddin, et al., Saudi Pharm. J. 21 (2013) 125-141. DOI:10.1016/j.jsps.2012.05.009 |
| [76] |
M. MoralesCruz, G.M. FloresFernández, M. MoralesCruz, et al., Results Pharma. Sci. 2 (2012) 79-85. DOI:10.1016/j.rinphs.2012.11.001 |
| [77] |
M. Li, O. Rouaud, D. Poncelet, Int. J. Pharm. 363 (2008) 26-39. DOI:10.1016/j.ijpharm.2008.07.018 |
| [78] |
M. Cho, H. Sah, J. Microencapsul. 22 (2005) 1-12. DOI:10.1080/02652040400026269 |
| [79] |
J. Liu, Y. Xu, Z. Liu, et al., Eur. J. Pharm. Biopharm. 144 (2019) 217-229. DOI:10.1016/j.ejpb.2019.09.022 |
| [80] |
D. Blanco, M.J. Alonso, Eur. J. Pharm. Biopharm. 45 (1998) 285-294. DOI:10.1016/S0939-6411(98)00011-3 |
| [81] |
D. Chitkara, N. Kumar, Pharm. Res. 30 (2013) 2396-2409. DOI:10.1007/s11095-013-1084-6 |
| [82] |
M.D. Blanco, M.J. Alonso, Eur. J. Pharm. Biopharm. 43 (1997) 287-294. DOI:10.1016/S0939-6411(97)00056-8 |
| [83] |
J. Cheng, B.A. Teply, I. Sherifi, et al., Biomaterials 28 (2007) 869-876. DOI:10.1016/j.biomaterials.2006.09.047 |
| [84] |
S. Fredenberg, M. Wahlgren, M. Reslow, A. Axelsson, Int. J. Pharm. 415 (2011) 34-52. DOI:10.1016/j.ijpharm.2011.05.049 |
| [85] |
M.A. Tracy, K.L. Ward, L. Firouzabadian, et al., Biomaterials 20 (1999) 1057-1062. DOI:10.1016/S0142-9612(99)00002-2 |
| [86] |
T.G. Park, Biomaterials 16 (1995) 1123-1130. DOI:10.1016/0142-9612(95)93575-X |
| [87] |
J.S. Wiggins, M.K. Hassan, K.A. Mauritz, R.F. Storey, Polymer 47 (2006) 1960-1969. DOI:10.1016/j.polymer.2006.01.021 |
| [88] |
J.V. Andhariya, R. Jog, J. Shen, et al., J. Control. Release 308 (2019) 1-13. DOI:10.1016/j.jconrel.2019.07.013 |
| [89] |
J. Liu, Y. Xu, Y. Wang, et al., Eur. J. Pharm. Sci. 134 (2019) 69-80. DOI:10.1016/j.ejps.2019.04.017 |
| [90] |
N. Guo, Q. Zhang, Y. Sun, H. Yang, Int. J. Pharm. 560 (2019) 273-281. DOI:10.1016/j.ijpharm.2019.01.061 |
| [91] |
R.D. Vaishya, A. Mandal, M. Gokulgandhi, S. Patel, A.K. Mitra, Int. J. Pharm. 489 (2015) 237-245. DOI:10.1016/j.ijpharm.2015.04.075 |
| [92] |
F. Qi, L. Yang, J. Wu, G. Ma, Z. Su, Pharm. Res. 32 (2015) 2310-2317. DOI:10.1007/s11095-015-1622-5 |
| [93] |
S.M. Lim, H.N. Eom, H.H. Jiang, M. Sohn, K.C. Lee, J. Pharm. Sci. 104 (2015) 72-80. DOI:10.1002/jps.24238 |
| [94] |
A. Giteau, M.C. VenierJulienne, A. AubertPouëssel, J.P. Benoit, Int. J. Pharm. 350 (2008) 14-26. DOI:10.1016/j.ijpharm.2007.11.012 |
| [95] |
M. Weert, J. Hoechstetter, W.E. Hennink, D.J.A. Crommelin, J. Control. Release 68 (2000) 351-359. DOI:10.1016/S0168-3659(00)00277-7 |
| [96] |
M. Nuzzo, A. MillqvistFureby, J. Sloth, B. Bergenstahl, Dry. Technol. 33 (2015) 757-767. DOI:10.1080/07373937.2014.990566 |
| [97] |
X. Hong, L. Wei, L. Ma, et al., Int. J. Nanomed. 8 (2013) 2433-2441. |
| [98] |
P. Tomar, N. Giri, V.S. Karwasara, R.S. Pandey, V.K. Dixit, Pharm. Dev. Technol. 17 (2012) 421-428. DOI:10.3109/10837450.2010.546408 |
| [99] |
S. Marquette, C. Peerboom, A. Yates, et al., Eur. J. Pharm. Biopharm. 86 (2014) 393-403. DOI:10.1016/j.ejpb.2013.10.013 |
| [100] |
J. He, H. Li, C. Liu, et al., J. Microencapsul. 32 (2015) 608-617. DOI:10.3109/02652048.2015.1065924 |
| [101] |
M. Allahyari, E. Mohit, Hum. Vaccin. Immunother. 12 (2016) 806-828. DOI:10.1080/21645515.2015.1102804 |
| [102] |
J. Siepmann, K. Elkharraz, F. Siepmann, D. Klose, Biomacromolecules 6 (2005) 2312-2319. DOI:10.1021/bm050228k |
| [103] |
C. Dai, B. Wang, H. Zhao, Colloids Surf. B: Biointerfaces 41 (2005) 117-120. DOI:10.1016/j.colsurfb.2004.10.032 |
| [104] |
F. Wan, M.J. Maltesen, S.K. Andersen, et al., Pharm. Res. 31 (2014) 2940-2951. DOI:10.1007/s11095-014-1387-2 |
| [105] |
J. Meeus, D.J. Scurr, K. Amssoms, et al., Mol. Pharm. 10 (2013) 3213-3224. DOI:10.1021/mp400263d |
| [106] |
Y. Wang, D.J. Burgess, Int. J. Pharm. 454 (2013) 310-315. DOI:10.1016/j.ijpharm.2013.06.012 |
| [107] |
T.A. Ahmed, Y.A. Alharby, A.R.M. ElHelw, K.M.H. Ibrahim, K.M. ElSay, Drug Des. Devel. Ther. 10 (2016) 405-415. |
| [108] |
H. Liu, S.S. Venkatraman, J. Biomater. Sci. Polym. Ed. 23 (2012) 251-266. DOI:10.1163/092050610X549171 |
| [109] |
A.N. Ford Versypt, D.W. Pack, R.D. Braatz, J. Control. Release 165 (2013) 29-37. DOI:10.1016/j.jconrel.2012.10.015 |
| [110] |
G. Mittal, D.K. Sahana, V. Bhardwaj, M.N.V. Ravi Kumar, J. Control. Release 119 (2007) 77-85. DOI:10.1016/j.jconrel.2007.01.016 |
| [111] |
Z. Ghalanbor, M. Körber, R. Bodmeier, Eur. J. Pharm. Biopharm. 85 (2013) 624-630. DOI:10.1016/j.ejpb.2013.03.031 |
| [112] |
A. Cossé, C. König, A. Lamprecht, K.G. Wagner, AAPS PharmSciTech 18 (2017) 15-26. DOI:10.1208/s12249-016-0548-5 |
| [113] |
N. Nihant, C. Grandfils, R. Jérôme, P. Teyssié, J. Control. Release 35 (1995) 117-125. DOI:10.1016/0168-3659(95)00026-5 |
| [114] |
X. Zheng, Z. Fang, W. Huang, et al., Acta Pharm. Sin. B 12 (2022) 3972-3985. DOI:10.1016/j.apsb.2022.04.011 |
| [115] |
W. Huang, Z. Fang, X. Zheng, et al., Chin. Chem. Lett. 33 (2022) 4079-4083. DOI:10.1016/j.cclet.2022.01.043 |
| [116] |
K. Liu, W. Liu, Z. Dong, et al., Bioeng. Transl. Med. (2022), https://aiche.onlinelbrary.wiley.com/doi/full/10.1002/btm2.10405.
|
| [117] |
Z. Li, H. Mu, S. Larsen, et al., Int. J. Pharm. 609 (2021) 121183. DOI:10.1016/j.ijpharm.2021.121183 |
| [118] |
C. Bassand, J. Verin, M. Lamatsch, et al., J. Control. Release 343 (2022) 255-266. DOI:10.1016/j.jconrel.2022.01.028 |
| [119] |
Y. Cai, J. Qi, Y. Lu, H. He, W. Wu, Adv. Drug Deliv. Rev. 118 (2022) 114463. |
| [120] |
H. He, C. Liu, J. Ming, et al., Aggregate 3 (2022) e163. DOI:10.1002/agt2.163 |
| [121] |
Y. Cai, X. Ji, Y. Zhang, et al., Aggregate (2022), https://onlinelibrary.wiley.com/doi/full/10.1002/agt2.277.
|
| [122] |
J. Bai, J. Zhou, X. Ji, et al., Chin. Chem. Lett. 33 (2022) 4175-4178. DOI:10.1016/j.cclet.2022.02.017 |
| [123] |
J. Yang, Z. Dong, W. Liu, et al., Chin. Chem. Lett. 31 (2020) 875-879. DOI:10.1016/j.cclet.2019.07.016 |
| [124] |
L. Hang, C. Shen, B. Shen, H. Yuan, Chin. Chem. Lett. 33 (2022) 4948-4951. DOI:10.1016/j.cclet.2022.03.108 |
| [125] |
D. Liu, B. Wan, J. Qi, et al., Chin. Chem. Lett. 29 (2018) 1834-1838. DOI:10.1016/j.cclet.2018.11.015 |