 2023, Vol. 34
2023, Vol. 34 


b CAMS Key Laboratory of Antiviral Drug Research,Beijing Key Laboratory of Antimicrobial Agents,NHC Key Laboratory of Biotechnology of Antibiotics,Institute of Medicinal Biotechnology,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100050,China
Hepatitis B virus (HBV),a significant cause of viral hepatitis in humans,belongs to the Hepadnaviridae family and has a double-stranded relaxed circular DNA (rcDNA) [1]. Globally,an estimated 257 million people are living with HBV infection despite the improving coverage of preventative vaccines [2]. Those chronically infected patients are more likely to progress to cirrhosis or even hepatocellular carcinoma,a leading cause of cancer-related death worldwide [3,4]. HBV infection remains a severe global public health problem.
Current therapeutic options,including nucleos(t)ide analogs (NAs) and peginterferon (PEG-IFN),can efficaciously suppress HBV replication,thus delaying the progression of liver diseases [5,6]. Nonetheless,these two available treatment modalities still suffer various limitations and rarely eventually achieve a cure or a functional cure. Long-term NA treatment leads to the emergence of drug-resistant HBV variants [7]. When it comes to interferon-based treatment,it has only a modest response rate,requires subcutaneous injection,close monitoring of treatment,high costs,and usually causes serious side effects [8]. Both NAs and PEG-IFN fail to eliminate covalently closed circular DNA (cccDNA) and integrated HBV DNA,which are the main culprits in viral persistence [9,10]. Therefore,it is imperative to develop safe and effective direct antiviral agents (DAAs) that can induce the loss or inactivation of cccDNA and achieve a functional cure or a cure [11].
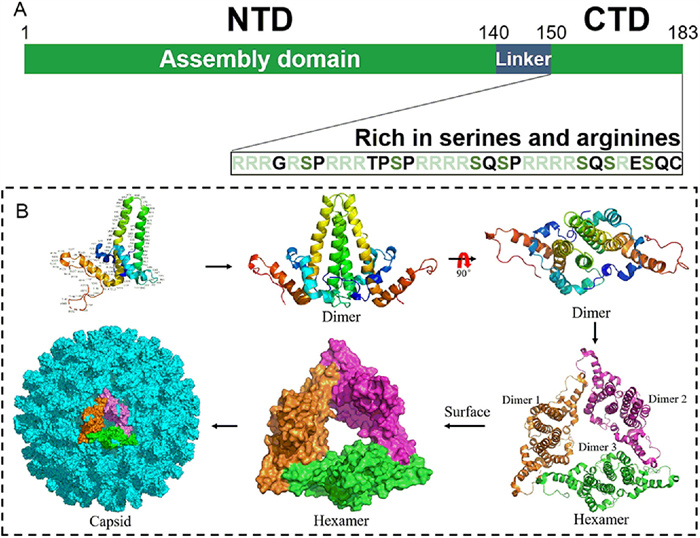
The discovery of novel small-molecule DAAs for different anti-HBV targets holds the promise of improving the effectiveness of existing treatments and avoiding or mitigating adverse effects. The HBV core protein (Cp),a well-conserved 183-residue protein,is an excellent target for antiviral agents (Fig. 1A) [12]. The N-terminal domain (NTD) consisting of the first 140 residues,acts as the assembly domain,which alone can self-assemble into empty capsids in vitro [13]. This domain comprises 5 α-helices connected by loops. The C-terminal domain (CTD),which is considered as nucleic acid chaperone,includes the last 34 residues and is rich in serines and arginines [14,15]. Phosphorylation and dephosphorylation in this domain were found to be associated with the specific packaging of pgRNA,intracellular transport of HBV Cps,capsid maturation,envelopment and secretion. Newly translated Cps firstly assemble into homodimers,among which three dimers interact with HBV pregenomic RNA (pgRNA) and polymerase to form a precursor hexamer (Fig. 1B) [16]. Subsequently,Cp dimers are rapidly added to this hexamer one at a time and eventually assemble into an intact capsid. The mature capsid is an icosahedral structure mostly formed by 120 dimers through hydrophobic interactions [17]. It involves in or modulates almost every stage of the HBV lifecycle,including the endocytosis of virion,nuclear trafficking,synthesis of cccDNA,reverse transcription,pgRNA packaging,recycling of the capsid,and the formation of mature virus particles (Fig. S1 in Supporting information),which makes it an attractive target [18]. The core protein allosteric modulators (CpAMs) can acutely impact the correct capsid assembly,thereby suppressing HBV replication. Interestingly,various CpAMs have also been demonstrated to inhibit the de novo synthesis of cccDNA when they were present during the early stages of the HBV replication cycle,though the already established cccDNA in cells could not be influenced [19-22].

|
Download:
|
| Fig. 1. (A) Domain structure of core protein and structural motifs at C-terminal domain. (B) A schematic of the HBV capsid assembly process. The crystal structures of HBV core protein monomer,dimer (PDB code: 1QGT),hexamer (PDB code: 5WRE) and capsid (PDB code: 7OD4) were generated in PyMOL (www.pymol.org). | |
{kind=link}
According to chemical structures,CpAMs can be divided into derivatives of heteroaryldihydropyrimidine (HAP),sulfamoylbenzamide (SBA),sulfamoylpyrroloamide (SPA),phenylpropenamide (PPA),benzamide (BA),pyridazinone and various other scaffolds [23]. They are grouped into two classes (Ⅰ and Ⅱ) depending on the mode of action (MoA) to interfere with capsid assembly [24,25]. Class Ⅰ molecules,including HAPs,lead to the formation of aberrant capsids or large aggregates of Cp,which are ultimately degraded within cells; whereas class Ⅱ molecules,such as SBAs and BAs,induce the formation of morphologically normal but pgRNA-free empty capsids. All CpAMs reported so far bind to the same hydrophobic pocket located at the dimer-dimer interface near the C-termini of the core assembly subunits [26-29]. In recent years,several CpAMs enter clinical trials (Table S1 in Supporting information). However,in spite of such successes,there are no approved CpAM-type drugs for the treatment of HBV infection to date,mainly due to adverse events (AEs) or insufficient efficacy when used alone [29]. Broadly speaking,compounds targeting capsid have also experienced high attrition rates in clinical trials,for example,in the field of anti-HIV drugs,although HIV capsid modulators have been studied for more than 20 years,it was only last year that the first drug came to market [30-33]. Therefore,iterative optimization of existing compounds and development of new skeletons are still essential.
Innovation of therapeutic drug entities and development of unconventional drug discovery technologies were two aspects of seeking HBV CpAMs. Consequently,in this paper,the latest research progress (2018–2022) of HBV CpAMs is reviewed from the perspective of medicinal chemistry strategies to guide further development of new and effective anti-HBV drugs.
2. Medicinal chemistry strategies in the recent discovery and optimization of HBV core protein allosteric modulators 2.1. Leads compound discoveryOver the last few years,new technological and scientific developments have been revolutionizing the drug discovery process. High-throughput (phenotypic) screening,virtual screening of compound libraries (synthetic or natural compounds),and drug repositioning are currently the main technologies used to identify new HBV CpAMs hit compounds.
2.1.1. High-throughput screeningSupported by information management software,high-throughput screening (HTS) automates the experimental operation,data acquisition,and analytical processing. Using HTS method,trace amount,high speed,and high screening volume can be achieved for the analysis of substantial biochemical substances in drug discovery [34]. It is widely employed to search for lead compounds,as exemplified by the recent discovery of HBV CpAMs.
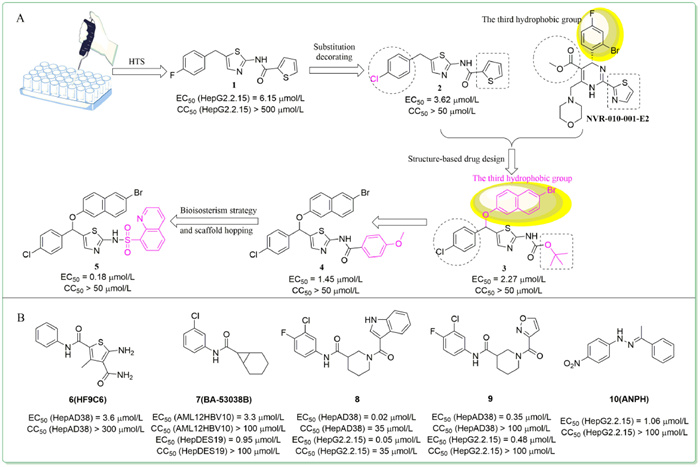
In 2019,HTS of a commercial drug-like chemical library conducted by Pan et al. obtained a hit compound 1 (Fig. 2A) with high potency and low cytotoxicity (EC50 = 6.15 µmol/L,CC50 > 500 µmol/L) [35]. Preliminary structure–activity relationship (SAR) studies showed that chlorine-substituted benzene derivatives were more active than fluorine and bromine,represented by compound 2. By referring to the successful HAP structure of NVR-010-001-E2,compound 3 with α-bromide-naphthalene moiety as the third hydrophobic group was then disclosed and displayed better inhibitory activity (EC50 = 2.27 µmol/L). The α-bromide-naphthalene group appeared to bind with the hydrophobic cavity of the capsid that was occupied by the halogenated benzene of NVR-010-001-E2. Subsequent structural modifications of compound 3 led to the discovery of 4 with an EC50 value of 1.45 µmol/L. To further optimize 4,sulfonamide series were tested and compound 5 featured by a quinoline group was obtained. 5 inhibited HBV replication both in vitro (EC50 = 0.18 µmol/L,CC50 > 50 µmol/L) and in vivo with no obvious acute toxicity,which made it attractive to be exploited in future drug development. However,it should not be ignored that 5 had a too high logP value for acceptable drug metabolism and pharmacokinetics (DMPK) properties (clogP = 9.06).

|
Download:
|
| Fig. 2. (A) HTS and SAR exploration in the development of 5; (B) The chemical structures of compounds 6–10 identified by HTS. | |
{kind=link}
In the same year,Huber et al. proposed a thermal shift-based HTS method for CpAM discovery and characterization [36]. Prompted by this innovative method,compound 6 (HF9C6) (Fig. 2B) with a benzamide scaffold was obtained and chosen for further studies as it showed the highest potency to inhibit HBV replication and low cytotoxicity in HepAD38 cells (EC50 = 3.6 µmol/L,CC50 > 300 µmol/L). Remarkably,a panel of mutants showed complementary resistance profiles between Bay 38-7690 and 6,which highlighted the possibility of combination therapies of multiple CpAMs. Another novel benzamide derivative 7 (BA-53038B) was also identified by HTS [37]. Compound 7 induced the formation of morphologically normal empty capsids with decreased electrophoresis mobility,while other class Ⅱ CpAMs examined so far with increased electrophoresis mobility,indicating that 7 exerted its effect in a distinct manner. Additionally,Pei et al. developed a simple and effective HTS assay using a HepAD38 luciferase reporter cell line and obtained another two novel benzamide derivatives,8 and 9 [38]. Mechanistic studies confirmed that they accelerated the formation of morphologically normal empty capsids,hence both were class Ⅱ CpAMs.
In 2021,Yamasaki et al. screened an in-house compound library using the HBV103-AdV system [39]. Compound 10 (ANPH) was selected for further characterization since its highest activity and low cytotoxicity (EC50 = 1.06 µmol/L,CC50 > 100 µmol/L). Mechanistic studies revealed that 10 was a class Ⅱ CpAM.
2.1.2. Virtual screeningHigh-throughput screening of large libraries of compounds is a time-consuming and expensive process. With the rapid development of computational chemistry and molecular simulation,virtual screening enables realistic in vitro operations to be converted to virtual in silico computing. Although virtual screening is impossible to replace true in vitro pharmacological screening,it has an irreplaceable role in accelerating new drug discovery and guiding rational drug design [40]. Strategy of virtual screening has boosted the discovery of new skeletons CpAMs in recent years.
For instance,in 2019,Toyama et al. reported a pyrimidotriazine derivative 11 (Fig. S2 in Supporting information) through in silico screening [41]. Using substitution decorating approach,compound 12 was synthesized and displayed higher anti-HBV activity (EC50 = 5.8 µmol/L,CC50 > 100 µmol/L). Compound 12 was proven to be a potential class Ⅰ CpAMs using a cell-free capsid assembly system.
In 2021,Senaweera et al. conducted structure-based virtual screening against a small molecule protein-protein interaction (PPI) library using the known co-crystal structure of Cp with HAP_R01 [42]. 100 compounds out of 40,640 were selected by virtual screening for further biological evaluation. Among them,compound 13,another benzamide derivative,showed the best but still insufficient anti-HBV activity and modest cytotoxicity (EC50 = 17.2 µmol/L,CC50 = 47 µmol/L). Additionally,employing structure-based virtual screening,Wang et al. found that compound 14 with 2-aryl-4-quinolyl amide scaffold displayed anti-HBV activity with an EC50 value of 5.6 µmol/L [43]. Further structural optimization of compound 14 led to the discovery of compound 15 with improved activity (EC50 = 1.8 µmol/L). As demonstrated in bimolecular fluorescence complementation (BiFC) assay,compound 15 acted as a HBV CpAM,significantly disrupting the capsid interactions.
In 2022,to identify novel chemotypes of CpAMs,Yang et al. conducted structure-based virtual screening of an integrated compound library and discovered a series of N-sulfonylpiperidine-3-carboxamides (SPCs) compounds that potently reduced the amount of secreted HBV DNA [44]. Based on further SAR studies and optimization of the representative compound 16,compound 17 which exhibited the best anti-HBV effect with an EC50 value of 0.056 µmol/L was identified. 17 remained sensitive to a nucleos(t)ide-resistant variant and showed synergistic antiviral activity with ETV in vitro,which indicated that combination therapies of CpAMs and NAs may be beneficial.
Unquestionably,contemporary medicinal computational chemistry requires the usage of more sophisticated approaches,including data science,artificial intelligence,molecular simulations through classical mechanics and quantum mechanics,and cheminformatics [45,46]. Such endeavors would greatly improve the success rate of capsid-targeted virtual screening.
2.1.3. Drug repositioningOld drugs have already been applied in clinical practice,so their physical and chemical properties,pharmacokinetics (PK) properties,and security are confirmed and guaranteed. It is well known that new drug development is a high-failure rate,high-cost and rather slow endeavor. Drug repositioning,on the other hand,has the advantage of low research and development (R & D) costs,short development timelines,and significantly reduces uncertainties in safety and pharmacokinetic research [47,48]. Consequently,drug repositioning strategy is becoming increasingly attractive in drug discovery [49]. The development of HBV CpAMs has no exception.
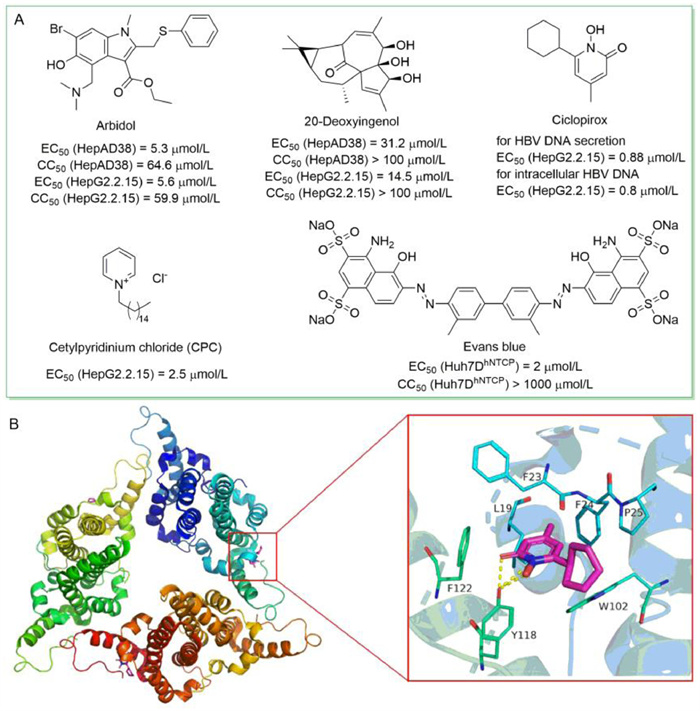
In 2018,Wei et al. developed a cell-based method for screening of compounds that targeting Cp dimerization,quantified by the split luciferase complementation (SLC) assay [50]. Prompted by this system,two libraries containing 304 natural products and 368 synthesized compounds were screened. Arbidol and 20-deoxyingenol (Fig. 3A) were identified and turned out to inhibit HBV replication in vitro by blocking or promoting the capsid formation process,respectively. Arbidol is a broad-spectrum antiviral drug [51],while 20-deoxyingenol is extracted from Caper Euphorbia Seed,a widely used traditional Chinese herbal medicine.

|
Download:
|
| Fig. 3. (A) The chemical structures of anti-HBV agents identified by drug repositioning. (B) The co-crystal structure of ciclopirox in complex with HBV core protein (PDB code: 6J10). The figure was generated in PyMOL (www.pymol.org). | |
{kind=link}
In 2019,Kang et al. screened 978 Food and Drug Administration (FDA)-approved drugs for their ability to inhibit HBV replication [52]. Ciclopirox,an antifungal drug,was found to strongly inhibit HBV replication both in vitro (EC50 = 0.88 µmol/L) and in mice by blocking capsid assembly. According to the protein-ligand co-crystal structure (Fig. 3B),there were extensive van der Waals interactions between ciclopirox and the binding pocket formed by hydrophobic residues L19,F23,F24,P25,Y118,F122 and W102. Specially,Y118 formed essential hydrogen bonds and salt bridges with carbonyl oxygen,nitrogen,and hydroxyl group of the pyridine ring of ciclopirox to anchor it in the binding pocket. Moreover,combination of ciclopirox and NAs showed synergistic effect. In conclusion,orally-administered ciclopirox has the potential to be developed into a promising treatment to combat HBV infection.
In the same year,Seo et al. identified cetylpyridinium chloride (CPC) as a novel CpAM with effective activity in vitro (EC50 = 2.5 µmol/L) and in vivo [53]. As a topical antibacterial agent in the oropharynx,CPC has been used as an adjunctive treatment for oral infectious diseases [54]. It has also been widely used in over-the-counter products for instance mouthwashes and dentifrices. Mechanistic studies confirmed that CPC interacted with the HBV Cp149 dimer and induced its conformational change. Moreover,CPC turned out to synergize with NAs to prevent HBV replication in a mouse model. In addition,Xiao et al. screened an FDA drug library of 1280 compounds using Huh7DhNTCP cells and identified Evans blue as a novel HBV inhibitor [55]. Evans blue displayed excellent activity and low cytotoxicity in Huh7DhNTCP cells (EC50 = 2 µmol/L,CC50 > 1000 µmol/L). Mechanistic studies showed that Evans blue had a dual anti-HBV effect,blocking both the HBV entry through inhibiting the binding of viral preS1 to the host factor NTCP (sodium taurocholate cotransporting polypeptide) and capsid assembly through targeting another host factor BKCa channel. Presumptively,Evans blue is less prone to emergence of resistance since it targets host factors. Moreover,Evans blue could inhibit the infection of NA-resistant HBV strains. Apparently,it is a promising anti-HBV drug candidate for further investigation.
In addition,virtual screening also represents a useful tool for performing effective drug repositioning for existing drugs in the market,with a remarkable reduction in the developmental costs of cell-based anti-HBV screening and target validation [56,57]. In conjunction with machine learning models,cutting-edge computational methodologies should be established to infer new drug activity and accelerate drug repositioning.
2.1.4. Natural productsThere exists an enormous structural complexity and diversity among secondary metabolites from natural products,which inspires medicinal chemists to dig into their infinite potential [58-60]. Recently there has been a renewed interest in natural products research due to the failure of alternative drug discovery methods to deliver lead compounds in major therapeutic areas including antiviral therapies. The bis-heterocycle compound NZ-4 was designed and identified based on the structural optimization of leucamide A,a cyclic heptapeptide isolated from the Australian marine sponge Leucetta microraphis [61,62]. More CpAMs obtained from natural products are worth expecting.
In 2022,Luo et al. found that the ethanol extract of R. japonicus Thunb. could inhibit HBV replication through screening a traditional Chinese medicine pool [63]. Mechanistic studies revealed that R. japonicus Thunb. extract functioned at multiple stages of the viral lifecycle,especially the processes of cccDNA formation and capsid assembly. The isolation and identification of active compounds and their structural modification are worthy of further efforts.
We envisioned that the intrinsic complexity of natural-product-based drug discovery necessitates highly integrated interdisciplinary approaches to continue making significant contributions to anti-HBV agents.
2.2. Optimization of lead compoundsLead compounds discovered by above-mentioned methods hardly reach the acme of perfection in terms of anti-HBV potency and druggability. Therefore,adopting various strategies to optimize them is necessary. Just as Nobel laureate (1988) Sir James Black said,"the most fruitful basis of the discovery of a new drug is to start with an old one."
2.2.1. Substitution decorating approachAs a basic lead optimization method,substitution decorating is commonly used in contemporary drug discovery to extract SAR information for further rational modifications,as well as to improve activity and drug-like properties [64-67]. In recent years,with existing CpAMs as lead compounds,some novel and more prominent derivatives were obtained using substitution decorating approach.
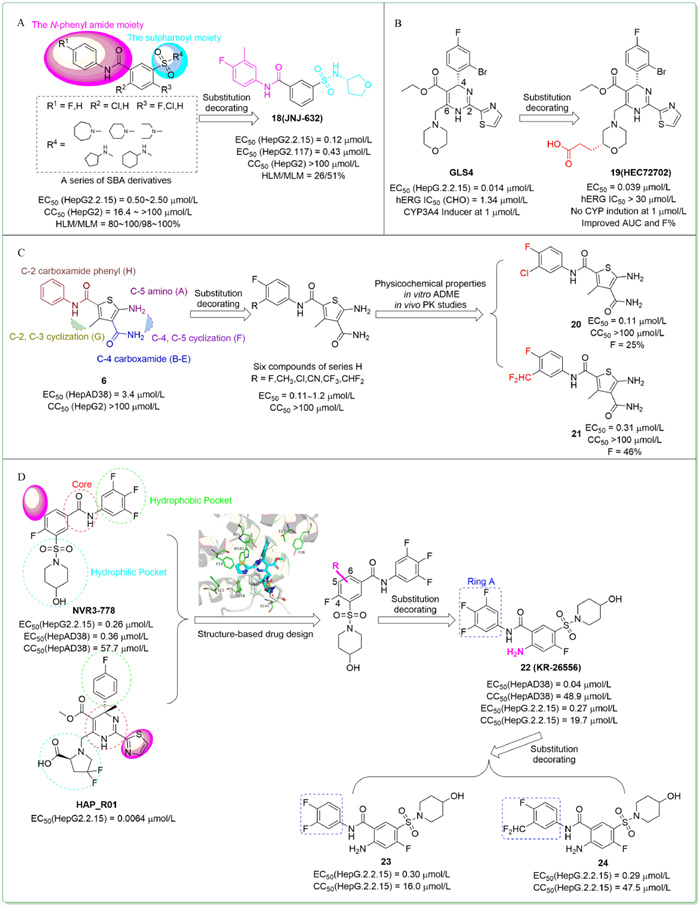
Aiming at reinforcing activity and enhancing metabolic stability,in 2018,Vandyck et al. rationally explored different substitution patterns of the sulphamoyl and N-phenyl amide moieties of a series of SBA derivatives [68]. Among these compounds,18 (JNJ-632) (Fig. 4A) was selected for further studies because it displayed high activity,weak cytotoxicity,and relatively superior metabolic stability (EC50 = 0.12 µmol/L in HepG2.2.15 cells,EC50 = 0.43 µmol/L in HepG2.117 cells,CC50 > 100 µmol/L in HepG2 cells,human/mouse liver microsome stability (expressed as percentage metabolization after 15 min of incubation) = 26/51%),which meant a suitable in vitro profile. In vivo studies also showed that subcutaneous administration of 18 in HBV-infected chimeric mice led to a marked reduction of the HBV DNA viral load.

|
Download:
|
| Fig. 4. The chemical structures of compounds 18–24 through substitution decorating approach. The co-crystal structure of core protein with NVR 3-778 (PDB code: 5T2P) and HAP_R01 (PDB code: 5WRE) shown in (A) was generated in PyMOL (www.pymol.org). | |
{kind=link}
In the same year,Ren et al. reported the design and synthesis of novel derivatives through substitution decorating of GLS4,in an attempt to find a better compound with reasonable antiviral potency,attenuated hERG activity,decreased CYP enzyme induction,and improved PK properties [69]. Firstly,replacing the dihydropyrimidine core with some other heterocycles led to significant drops in anti-HBV potency,though decreased hERG inhibitions. Subsequently,the team modified the structure of the C-2,C-4 and C-6 positions while keeping the core unchanged to obtain another three series of derivatives. Considering the possibly important role of chirality for antiviral potency,compounds with good anti-HBV activities and low hERG activities obtained above were chirally separated to further study the chiral isomers. The obtained compound 19 (HEC72702) (Fig. 4B),with an (R)morpholine-2-propionic acid at the C-6 position displayed the best overall profile in in vitro potency (EC50 = 0.039 µmol/L),in vitro metabolic stability,PK properties,hERG inhibition,and CYP enzymes induction.
In 2019,Tang et al. optimized the previously found benzamide derivative 6 through substitution decorating approach [70]. They explored the SAR of five substitutions around the central thiophene skeleton and designed eight series of compounds,series A–H. Among them,six compounds of series H,which bear a para F atom and a meta substitution at the aromatic ring,displayed significantly enhanced anti-HBV activity (EC50 = 0.11–1.2 µmol/L) and no cytotoxicity up to 100 µmol/L. To further assess their drug-like properties,physicochemical properties,in vitro ADME,and in vivo PK studies were conducted. Two of analogs,20 (EC50 = 0.11 µmol/L,CC50 > 100 µmol/L,F = 25%) and 21 (EC50 = 0.31 µmol/L,CC50 > 100 µmol/L,F = 46%) (Fig. 4C) exhibited overall superior profiles in these studies,indicating that both were high potential candidates for preclinical development.
In 2020,Na et al. reported the design and synthesis of novel SBA derivatives through substitution decorating of NVR3-778 [71]. The superimposed co-crystal structures of NVR3-778 and HAP_R01 reflected that most binding interactions were similar,but the thiazole moiety at C-2 position of HAP_R01 could afford additional interactions with the capsid,which might be the reason for the higher potency of HAP_R01. Therefore,they performed substitution decorating at C-4,C-5 and C-6 positions of the benzamide core of NVR3-778 to improve its affinity. Finally,the substitution decorating at C-6 position resulted in the SBA derivative 22 (KR-26556) (Fig. 4D) with an EC50 value of 0.04 µmol/L,9-fold more potent than NVR3-778 in HepAD38 cells. 22 also showed a promising overall profile in safety assessment,metabolic stability assessment,in vitro and in vivo PK studies. Therefore,this group further investigated the SAR of the A ring of 22 and thus obtained compounds 23 and 24 with good anti-HBV activity and validated PK properties [72].
In 2021,Kim et al. reported a series of pyrimidine derivatives as HBV CpAMs. Starting from hit compound 25 (Fig. S3A in Supporting information),which was identified by screening of an in-house compound library,analogs with modifications to three substitutions around the central pyrimidine skeleton were synthesized and evaluated [73]. Several rounds of SAR studies resulted in the identification of 26,with an optimal profile in in vitro (EC50 = 0.181 µmol/L) and in vivo activity,in vitro metabolic stability,PK properties,Ames and hERG channel assays. Additionally,combination of 26 and tenofovir showed synergistic repression of HBV replication in human liver-chimeric uPA/SCID mice.
In 2022,Spunde et al. reported a new HAP compound 27 (Fig. S3B in Supporting information) through substitution decorating of Bay 41-4109 in C-5 position [74]. Among those compounds with substitution decorating in C-2,C-4,C-5,C-6 positions or the dihydropyrimidine core,27 displayed the highest potency and the lowest cytotoxicity (EC50 = 6.24 µmol/L,CC50 = 175 µmol/L). However,though the cytotoxicity of 27 was much lower than that of Bay 41-4109,its activity and selectivity index (SI) did not exceed that of the lead. Mechanistic studies showed that 27 induced the formation of aberrant capsids as well as the capsid aggregates,just like other class Ⅰ CpAMs.
In the same year,Lv et al. explored different substitution patterns of the linker,ring B,and ring C moieties of GYH2-18,a class Ⅱ CpAM with 6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxamine (DPPC) skeleton [75]. Among three series of compounds,five compounds 28–32 (Fig. S3C in Supporting information) were selected and further studied because of their high potency (EC50 = 0.008–0.016 µmol/L) and low cytotoxicity (CC50 > 200 µmol/L). All of them showed good oral PK properties,while 28 and 29 were superior to the lead GYH2-18. In order to determine influence of the chirality on anti-HBV potency,28,29 and GYH2-18 were chirally separated to further study the chiral isomers. It was found that (6S)-cyclopropyl DPPC isomers (EC50 = 0.009–0.020 µmol/L) displayed significantly higher potency than the corresponding (6R)-ones (EC50 = 0.109–0.866 µmol/L).
More recently,Kuduk et al. have reported the further optimization of a series of diazepinone CpAMs using unsubstituted 33 as the lead [76]. SAR exploration of the 8-position of the diazepinone led to the discovery of compound 34 (EC50 = 0.004 µmol/L) (Fig. S3D in Supporting information),which was 17-fold more active than 6 and maintained similar human microsomal clearance (CL = 12 mL min−1 kg−1) and good solubility (153 µmol/L at pH 7). Subsequent modifications focused on the urea moiety and thus obtained compound 35 with equal potency to 34. In order to understand what led to the significant increase in anti-HBV activity after the addition of a single fluorine atom,the co-crystal structure of the Cp Y132A mutant bound to 35 was solved. However,no essential interactions that could account for the increase in anti-HBV activity of the fluorine with the protein were found. One presumption is that the fluorine renders the adjacent methylene more acidic,resulting in reinforced interaction between the methylene group and the adjacent carbonyl of Leu140. Transforming to the cyano,fluoro phenyl urea,in company with the previously reported beneficial 6-Me substituent on the piperidine moiety,was explored and obtained compound 36,the (6R,8′R) isomer [77]. 36 displayed excellent cellular potency (EC50 = 0.0016 µmol/L),good solubility (72 µmol/L at pH 7),and desirable PK profiles.
The high hit rate and the potency of the new CpAMs demonstrated the efficiency of the substitution decorating approach around the privileged structure.
2.2.2. Scaffold hoppingScaffold hopping,a classical medicinal chemistry strategy,typically starts with known active compounds and ends with novel scaffold compounds through transformation of the parental nucleus structure [78]. It can be conducted through computational methods as well as on a rationally designed case-by-case basis. This strategy is extensively utilized by medicinal chemists to enhance activity,optimize druggability or obtain intellectual property (IP) based on the structure of lead compounds [79-81].
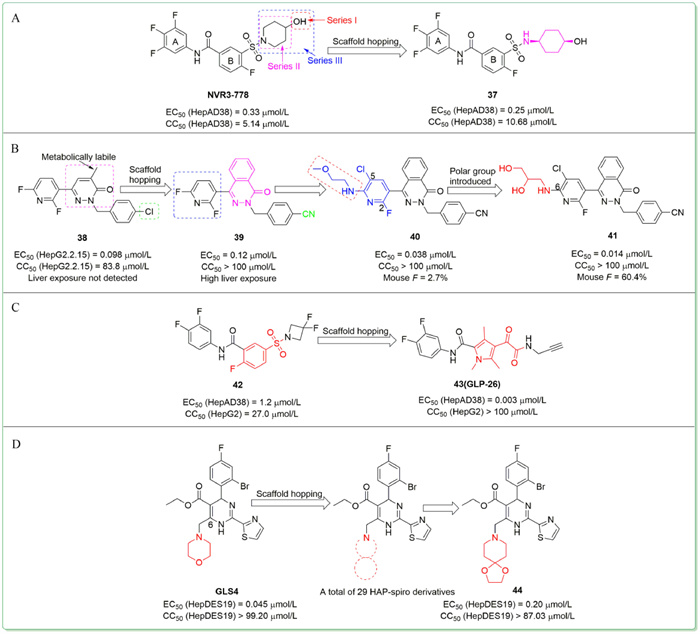
For example,in 2020,Lv et al. reported the design and synthesis of novel SBA derivatives using NVR3-778 as the lead compound [82]. Based on previous SAR studies and the reported crystal structure of Cp bound to NVR3-778,the team chose to modify the 4-hydoxyl-piperidyl moiety of NVR3-778 employing scaffold hopping and substitution decorating methods while keeping the ring A,B,and the benzamide moiety unchanged. Among three series of compounds,37 (Fig. 5A) stood out with potent anti-HBV activity,low cytotoxicity (EC50 = 0.25 µmol/L,CC50 = 10.68 µmol/L),and acceptable PK profiles.

|
Download:
|
| Fig. 5. The chemical structures of compounds 37–44 through scaffold hopping approach. | |
{kind=link}
In the same year,Chen et al. conducted SAR exploration of the previously identified compound 38 (Fig. 5B) based on its metabolic pathway study [83]. Firstly,to block the metabolic site of the methyl on the 4-methylpyridazinone moiety,scaffold hopping on this moiety resulted in compound 39 with moderate anti-HBV activity,low cytotoxicity (EC50 = 0.12 µmol/L,CC50 > 100 µmol/L) and improved liver distribution in ICR (CD-1) mice. Further optimization of the pyridine moiety was carried out while keeping fluorine substitution at the 2-position and thus 5-chloro substituted compound 40 with superior activity and low cytotoxicity was identified (EC50 = 0.038 µmol/L,CC50 > 100 µmol/L). To adjust the physical property of 40,the team kept 5-chloro substitution unchanged and explored diverse side chains at the 6-position of pyridine. Among them,41 bearing two hydroxy groups displayed an excellent antiviral potency with an EC50 value of 0.014 µmol/L and CC50 > 100 µmol/L. 41 also exhibited desirable drug-like properties with excellent oral bioavailability (F = 60.4%) in mice. Mechanistic studies showed that compound 41 could induce the formation of pgRNA-free empty capsids,hence it was a class Ⅱ CpAM.
In 2021,Amblard et al. reported the identification of compound 43 (GLP-26) (Fig. 5C),a glyoxamide derivative currently in preclinical studies [84,85]. Starting from a SBA derivative 42,preliminary in silico evaluation of the diketopyrole scaffold showed excellent similarity between the diketopyrole moiety and the benzyl-sulfonamide motif of 42. Thus,employing scaffold hopping strategy,this team designed and synthesized a series of diketopyrole derivatives,among which 43 was identified as the most promising compound with high potency and low cytotoxicity (EC50 = 0.003 µmol/L,CC50 > 100 µmol/L).
Spirocycles,with the right degree of conformational rigidity,high degree of saturation (expressed as Fsp3),and a pronounced three-dimensionality,represent an emerging privileged platform for drug design. Consequently,in continuation of our work on the HBV CpAMs,we reported the design and synthesis of 29 HAP-spiro derivatives [86]. The morpholine moiety at 6-position is located in a solvent-exposed region. Scaffold hopping strategy was adopted to replace the morpholine moiety of GLS4 with diverse spiro rings,in an attempt to improve the hydrophilicity and PK properties of GLS4. Among them,compound 44 (Fig. 5D) showed the best anti-HBV activity and low cytotoxicity,but did not exceed GLS4. Meanwhile,the predicted ADMET and PK profiles of 44 retained at the similar level as GLS4.
Additionally,Senaweera et al. carried out hit expansion via pharmacophore-based scaffold hopping approach [42]. Firstly,the common pharmacophore features of 45 (Janssen),46 (ZW-935) and 43 (GLP-26) were identified through in silico pharmacophore modeling. Subsequently,a series of compounds preserving these features were designed and selected according to their molecular docking scores and binding modes with key residues. Among them,two benzamide derivatives,47 and 48 (Fig. S4A in Supporting information) were disclosed as novel HBV CpAM hits with single-digit µmol/L EC50 and cytotoxicity up to 100 µmol/L. In early ADME assessment,both hits showed excellent plasma and microsomal stability in vitro. However,the aqueous solubility for both hits was less than ideal.
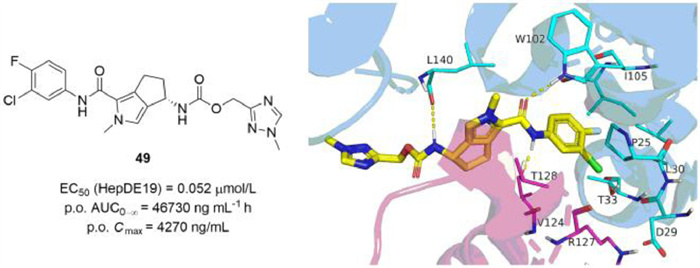
In 2022,Cole et al. reported the design and synthesis of derivatives through scaffold hopping of AB-506,a class Ⅱ molecule terminated in clinical phase I for its hepatotoxicity [87]. Based on the structural comparison of NVR 3-778 and JNJ56136379,it could be hypothesized that 1-methyl pyrrole is a tolerated replacement for the halogenated phenyl. Therefore,two enantiomers bearing bicyclic 1-methyl pyrrole heterocyclic were synthesized and evaluated. The (S)-enantiomer 49 (EC50 = 0.052 µmol/L) (Fig. S4B in Supporting information) displayed much better anti-HBV activity than its (R)-enantiomer (EC50 = 3.5 µmol/L),which was consistent with the lead AB-506. The co-crystal structure of the Cp Y132A mutant in complex with 49 was solved and the detailed interactions were provided (Fig. 6). Consistent with other published CpAMs,49 binds to the hydrophobic pocket located at the dimer-dimer interface near the C-termini of the core assembly subunits. The halogenated phenyl moiety inserts into a deep hydrophobic pocket formed by P25,D29,L30,T33,and I105,as well as V124 and R127 from another dimer. The secondary amide forms key hydrogen bonds with W102 and T128,while the (S)-enantiomer 49 forms an additional hydrogen bond with L140. In addition,the methyl triazole moiety is located at the edge of the binding site and is moderately solvent exposed. The minor modifications in this moiety had little influence on anti-HBV activity and led to the discovery of compounds 50 (EC50 = 0.050 µmol/L) and 51 (EC50 = 0.057 µmol/L) with similar activity and desirable PK profiles. The potent activity as well as high and sustained oral exposure of compounds 49,50 and 51 made a low once-daily dosing schedule possible.

|
Download:
|
| Fig. 6. The chemical structure of compound 49 through scaffold hopping approach and the co-crystal structure of core protein with compound 49 (PDB: 7S76),which was generated in PyMOL (www.pymol.org). | |
{kind=link}
Molecular hybridization involves the combination of pharmacophores from two different biologically active compounds in order to obtain a structurally novel compound with generally superior efficacy and desirable drug-like properties [88]. This effective approach has also been applied to the development of HBV CpAMs.
In 2020,we reported the design and synthesis of six series of heterocycle derivatives through combining scaffold hopping,bioisosterism,and molecular hybridization approaches,starting from NZ-4 and AT-130 [89]. There were compound 52 (Fig. S5A in Supporting information) possessed the best activity for anti-HBV DNA replication (EC50 = 2.2 µmol/L,CC50 = 80.8 µmol/L),and compounds 53 as well as 54 had the best activity for anti-HBsAg secretion and anti-HBeAg secretion,respectively. Moreover,surface plasmon resonance (SPR) analysis showed that the affinity constant of compound 54 (KD = 60.0 µmol/L) was comparable to that of the lead NZ-4 (KD = 50.6 µmol/L).
In 2022,Li et al. reported the discovery of a highly potent CpAM bearing a novel fused heterocycle amide scaffold,compound 60 (SHR5133) (Fig. S5B in Supporting information) currently in preclinical phase [90]. Initial hybridization between compounds 55 and 56 led to the identification of compound 57 containing the bicycle and amide combination with an EC50 value of 2.79 µmol/L. Then this team performed scaffold hopping of the bicycle,which afforded compound 58 with better activity (EC50 = 0.511 µmol/L). Further SAR studies on the imidazole scaffold and substitution of the piperazine disclosed two highly potent compounds 59 and 60. 60 showed better PK properties in rat than 59. In addition,it performed desirable PK profiles across species,favorable physicochemical properties,no CYP450 inhibition,and a high cardiac safety margin. Obviously,60 is a promising drug candidate worthy of further studies.
In the same year,Liu et al. used NVR3-778 and BA-38017 as lead compounds to design and synthesize three series of compounds [91]. Series Ⅰ and Ⅱ were designed based on the molecular hybridization of NVR3-778 and BA-38017,while scaffold hopping of the dihydrobenzodioxine moiety of BA-38017 resulted in series Ⅲ. Among these compounds,compound 61 (Fig. S6A in Supporting information) of series Ⅰ showed the best anti-HBV DNA replication activity and moderate cytotoxicity in HepAD38 cells (EC50 = 0.50 µmol/L,CC50 = 48.16 µmol/L).
Additionally,to address limitations of NVR3-778,including the modest antiviral activity and relatively low aqueous solubility,our team reported the design and synthesis of novel SBA derivatives using molecular hybridization and bioisosterism approaches [92]. Based on the comparison and analysis of co-crystal structure of HBV Cp Y132A mutant with NVR 3-778 and HAP_R01,we presume that the higher activity of HAP_R01 may be due to the additional hydrogen bond interactions of the carboxyl acid group with S141 at the solvent-exposed region. Therefore,we replaced the 4-hydroxyl-piperidine moiety of NVR 3-778 with carboxyl acids and obtained series I,in an attempt to enhance the interaction force with amino acids in the binding site as well as improve water solubility. Moreover,using bioisosterism strategy,series Ⅱ and Ⅲ bearing phenylboronic acids and phenylboronate esters were designed. Among three series of compounds,62 (Fig. S6B in Supporting information) showed comparable anti-HBV potency to NVR3-778 with better aqueous solubility. However,it is a pity that the bioavailability of 62 was only 2.85%,which motivates us to make further efforts.
It might be expected that availability of more precise protein models based on the X-ray crystal structures of ligands bound to HBV capsid could provide further valuable insights for structure-based molecular hybridization to facilitate hit-to-lead or lead-to-candidate development [93].
2.2.4. Prodrug strategyMany pharmacologically active agents with high potency have fatal flaws in drug-likeness due to poor solubility,deficient oral bioavailability,or low metabolic stability,which restrict their clinical applications seriously. The concept of "Prodrug" revives quite a few of them through simple but subtle modification [94]. An estimated 10% of drugs approved worldwide belong to the category of prodrugs [95]. Some attempts to take advantage of this concept also have been made in HBV CpAMs.
With the view of improving the water solubility of NVR 3-778,we reported the design and synthesis of four carboxylic acid and phosphate compounds based on NVR 3-778 [96]. There was no significant difference in anti-HBV activity between NVR 3-778 (EC50 = 0.38 µmol/L) and four prodrugs (EC50 = 0.42–0.28 µmol/L). Notably,compound 63 (Fig. S7 in Supporting information) not only improved the water solubility and released active drug more slowly in rat,but also increased the metabolic stability (63: T1/2 = 1.92 h; NVR 3-778: T1/2 = 1.40 h) in vitro. Moreover,the cytotoxicity of compound 64 (CC50 > 256 µmol/L) was significantly reduced compared with NVR 3-778 (CC50 = 13.65 µmol/L). Overall,it is a successful trial to improve drug-likeness of NVR3-778 through prodrug strategy.
2.2.5. Conformational constraint strategyTo fit with the conformation of the receptor binding site,a flexible ligand has to modulate its orientation and distance of groups to transform to the "active conformation" and this process leads to entropy loss. The conformational constraint of the flexible ligand can minimize this entropy loss,thus enhancing its affinity to the specific drug target [97]. It also has the potential to improve selectivity and metabolic stability [98]. Conformational constraint strategy plays an indispensable role in drug discovery and is commonly used by medicinal chemists to optimize lead compounds.
In 2020,Wang et al. adopted this strategy to discover compound 65 (Fig. S8 in Supporting information) using NVR3-778 as the lead compound [99]. In the active conformation of SBAs,the oxygen atom of the amide and carbon a in the figure are spatially close. To restrict this conformation,a new scaffold in which the C=O was replaced by the N atom in an aromatic heterocycle cyclized with carbon a was designed so that the N atom could form a hydrogen bond with W102 in a tight conformation. Combining with subsequent SAR studies on the indazole ring and other substitutions on the parent scaffold,compound 65 with excellent potency and low cytotoxicity was identified (EC50 = 0.034 µmol/L,CC50 > 100 µmol/L). 65 also displayed good metabolic stability in mouse liver microsomes but had the defect of poor aqueous solubility. Therefore,the team designed a series of amino acid prodrugs of 65,among which a citric acid salt 66 showed good solubility and demonstrated effectiveness in a hydrodynamic injection-based mouse model.
3. Conclusion and perspectivesThis mini-review provides a short overview of the function and structure of HBV core protein and summarizes the latest research progress (2018–2022) of HBV CpAMs. The typical medicinal chemistry strategies employed in the discovery and optimization of lead compounds were pointed out. Research paradigms and lessons learned from the discovery and development of HBV CpAMs will facilitate the discovery of additional drugs that specifically target various DNA and RNA capsids or other structural proteins.
Notably,high-throughput screening and virtual screening of compound libraries play an important role in the recent discovery of new structure HBV CpAMs,which owe to the continuous advance in computational chemistry. When it comes to drug repositioning,high-throughput phenotypic screening or inverse virtual screening for new indications of marketed or unapproved drugs is a highly efficient method. Natural products from animals,plants,microorganisms and marine organisms are also a nonnegligible source of new skeleton HBV CpAMs. Other drug discovery methods,such as fragment-based lead discovery and the application of DNA encoded libraries (DELs),hold potential as yet unexplored alternatives to expand the chemotypes amenable for development of HBV CpAMs [100,101].
Based on the reported chemical structures of CpAMs,ligand-based drug design strategies,such as bioisosterism,scaffold hopping,molecular hybridization,prodrug,and conformational constraint,have been employed to enrich the SAR information of the corresponding scaffolds,which facilitates further modifications to enhance antiviral activity and improve drug-like properties. With the development of structural biology,the crystal structures of ligands in complex with capsids have been solved,which boost the use of rational structure-based drug design. Targeting key amino acid residues to form additional interactions can lead to more efficient and effective optimization of lead compounds. Besides,to take full advantage of the wealth of structural biology information of HBV capsid,cheminformatics approach for intelligently designed small molecule library should be developed to maximize the diversity library realization.
HBV capsid shows high sequence conservation level. It plays an indispensable role in multiple stages of the HBV replication cycle. The assembly dynamics of capsid is so precise that even small interferences with the hydrophobic interaction between dimers of Cp can affect normal capsid assembly,thus disrupting the HBV lifecycle [102]. These factors impel CpAMs to become one of the most studied and promising anti-HBV candidates. However,the path to success will certainly be fraught with pitfalls and challenges (Fig. S9 in Supporting information). As discussed above,during the optimization of lead compounds,it is always difficult to balance the relationship between activity and druggability (including physicochemical properties,biochemical properties,pharmacokinetics properties and toxicity),as a result attending to one thing and losing another,which is a common problem in drug development. Secondly,although the establishment of the SARs of various scaffolds,the understanding of these SARs is usually not deep enough,and in many cases,we know the result but do not know the reason. In addition,there are concerns about the problem of drug resistance. Phase I proof-of-concept clinical trial of AB-506 reported a CHB patient who developed a natural mutation in Cp I105T at the baseline of treatment and thus presented a non-response status to a 160 mg/d dose of AB-506 [103]. This is the only reported case of primary resistance to CpAMs to date. But it can be foreseen that,with the extensive deepening of clinical research on CpAMs,more primary and secondary drug resistance mutations will be discovered.
To overcome these challenges,the multidisciplinary coordination between classical medicinal chemistry,structural biology,and computational chemistry is still necessary. Additionally,we need to pay more attention to the synchronous optimization of activity and druggability,and timely carry out the reciprocating feedback of "molecular design–chemical synthesis–multilevel biological evaluation". The parameters such as ligand efficiency (LE) and ligand lipophilicity efficiency (LLE) can be used to guide this process,which allows the combination of in vitro activity and physical and chemical properties to evaluate the quality of compounds [104,105]. Notably,solvent-exposed regions of HBV core proteins provide opportunities for substantial modifications of existing small-molecular drug molecules to regulate some physiochemical properties (cLogP,cLogD and Fsp3) that are associated with compound attrition,without serious loss of activity [106,107]. Experimental methods including isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) could be employed to determine the thermodynamic behavior of ligand binding to HBV capsid. The changes of free energy,enthalpy,and entropy are combined with the transformation of the complex structure to explain or reveal the SAR and guide the further optimization of compounds.
Introducing some advanced strategies in this field is also worth looking forward to. Proteolysis-targeting chimera (PROTAC) is a bifunctional molecule containing a ligand (mostly small-molecule inhibitor) of the protein of interest (POI) and a covalently linked ligand of the E3 ubiquitin ligase [108]. The small-molecule inhibitor recruits POI,while the ligand of E3 recruits the ubiquitin ligase,thus making the ubiquitinated protein degraded by the proteasome. Application of PROTAC strategy holds promise to achieve targeted degradation of the capsids and destroy their structure and function. Most interestingly,according to a proof-of-concept study,PROTAC molecule DGY-08-097 exhibited antiviral activity against mutant viruses that are resistant to the parental HCV protease inhibitor,which highlights the possibility of addressing viral drug resistance through PROTAC strategy [109]. Therefore,combining this strategy with CpAMs has a unique superiority and a good prospect. Besides the proteolytic machinery via the proteasomal system,lysosome-targeting chimaeras (LYTACs) and autophagy-targeting chimeras (AUTACs) trigger protein degradation via the lysosome and autophagy,separately. The rapid development of molecular glues and protein degraders with other protein degradation modalities expands their potential applications in HBV capsid [110]. Considering that capsid is structural protein with a broad and relatively superficial binding surface,the development of polyvalent CpAMs may improve binding affinity greatly compared with monovalent counterparts. Additionally,covalent inhibitors have the advantages of high potency,long effective action time,and good PK properties. Despite the promise that covalent-based antiviral therapeutics hold,it has remained a largely underexplored modality to develop covalent CpAMs [111]. All in all,these ideas here described may represent the blueprint for the future development of new generations of CpAM-based anti-HBV drugs.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC Nos. 82173677,82211530493),the Science Foundation for Outstanding Young Scholars of Shandong Province (No. ZR2020JQ31).
Supplementary materialsSupplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108349.
| [1] |
T.J. Liang, Hepatol. Baltim. Md. 49 (2009) S13-S21. DOI:10.1002/hep.22881 |
| [2] |
Global Hepatitis Report 2017,World Health Organization,19 April 2017,https://www.who.int/publications/i/item/9789241565455.
|
| [3] |
Y. Niu,W. Xie,W. Qin, Acta Pharm. Sin. B 1 (2011) 73-79. DOI:10.1016/j.apsb.2011.06.001 |
| [4] |
J. Balogh,D. Victor,E.H. Asham,et al., J. Hepatocell. Carcinoma 3 (2016) 41-53. DOI:10.2147/JHC.S61146 |
| [5] |
T. Asselah,D. Loureiro,N. Boyer,A. Mansouri, Lancet Gastroenterol. Hepatol. 4 (2019) 883-892. DOI:10.1016/S2468-1253(19)30190-6 |
| [6] |
M.H. Nguyen,G. Wong,E. Gane,et al., Clin. Microbiol. Rev. 33 (2020) e00046-19. |
| [7] |
J. Cheng,Y. Han,J.D. Jiang, Acta Pharm. Sin. B 4 (2014) 270-276. DOI:10.1016/j.apsb.2014.06.009 |
| [8] |
S. Fung,H.S.J. Choi,A. Gehring,H.L.A. Janssen, Hepatology 76 (2022) 233-250. DOI:10.1002/hep.32314 |
| [9] |
G.C. Fanning,F. Zoulim,J. Hou,A. Bertoletti, Nat. Rev. Drug Discov. 18 (2019) 827-844. DOI:10.1038/s41573-019-0037-0 |
| [10] |
J. Chang,F. Guo,X. Zhao,J.T. Guo, Acta Pharm. Sin. B 4 (2014) 248-257. DOI:10.1016/j.apsb.2014.05.002 |
| [11] |
Y. Pei,C. Wang,S.F. Yan,G. Liu, J. Med. Chem. 60 (2017) 6461-6479. DOI:10.1021/acs.jmedchem.6b01442 |
| [12] |
B.M. Chain,R. Myers, BMC Microbiol. 5 (2005) 33. DOI:10.1186/1471-2180-5-33 |
| [13] |
P.T. Wingfield,S.J. Stahl,R.W. Williams,A.C. Steven, Biochemistry 34 (1995) 4919-4932. DOI:10.1021/bi00015a003 |
| [14] |
M. Nassal, J. Virol. 66 (1992) 4107-4116. DOI:10.1128/jvi.66.7.4107-4116.1992 |
| [15] |
A. Diab,A. Foca,F. Zoulim,D. Durantel,O. Andrisani, Antiviral Res. 149 (2018) 211-220. DOI:10.1016/j.antiviral.2017.11.015 |
| [16] |
A. Zlotnick,J.M. Johnson,P.W. Wingfield,S.J. Stahl,D. Endres, Biochemistry 38 (1999) 14644-14652. DOI:10.1021/bi991611a |
| [17] |
A. Zlotnick,B. Venkatakrishnan,Z. Tan,et al., Antiviral Res. 121 (2015) 82-93. DOI:10.1016/j.antiviral.2015.06.020 |
| [18] |
U. Viswanathan,N. Mani,Z. Hu,et al., Antiviral Res. 182 (2020) 104917. DOI:10.1016/j.antiviral.2020.104917 |
| [19] |
F. Guo,Q. Zhao,M. Sheraz,et al., PLoS Pathog. 13 (2017) e1006658. DOI:10.1371/journal.ppat.1006658 |
| [20] |
J.M. Berke,P. Dehertogh,K. Vergauwen,et al., Antimicrob. Agents Chemother. 61 (2017) e00560-17. DOI:10.1128/AAC.00560-17 |
| [21] |
T. Lahlali,J.M. Berke,K. Vergauwen,et al., Antimicrob. Agents Chemother. 62 (2018) e00835-18. |
| [22] |
D. Burdette,A. Hyrina,Z. Song,et al., Antimicrob. Agents Chemother. 67 (2023) e01348-22. |
| [23] |
Y. Wang,S. Wang,X. Tao,et al., Med. Chem. Res. 31 (2022) 1414-1430. DOI:10.1007/s00044-022-02936-5 |
| [24] |
H. Kim,C. Ko,J.Y. Lee,M. Kim, Molecules 26 (2021) 7420. DOI:10.3390/molecules26247420 |
| [25] |
L. Yang,F. Liu,X. Tong,et al., ACS Infect. Dis. 5 (2019) 713-724. DOI:10.1021/acsinfecdis.8b00337 |
| [26] |
Z. Qiu,X. Lin,M. Zhou,et al., J. Med. Chem. 59 (2016) 7651-7666. DOI:10.1021/acs.jmedchem.6b00879 |
| [27] |
Z. Zhou,T. Hu,X. Zhou,et al., Sci. Rep. 7 (2017) 42374. DOI:10.1038/srep42374 |
| [28] |
S.P. Katen,Z. Tan,S.R. Chirapu,M.G. Finn,A. Zlotnick, Structure 21 (2013) 1406-1416. DOI:10.1016/j.str.2013.06.013 |
| [29] |
V. Taverniti,G. Ligat,Y. Debing,et al., J. Clin. Med. 11 (2022) 1349. DOI:10.3390/jcm11051349 |
| [30] |
D. Ding,S. Xu,X. Zhang,et al., Expert Opin. Drug Discov. 18 (2023) 5-12. DOI:10.1080/17460441.2023.2157401 |
| [31] |
X. Zhang,S. Xu,L. Sun,et al., Future Med. Chem. 14 (2022) 605-607. DOI:10.4155/fmc-2022-0008 |
| [32] |
L. Sun,X. Zhang,S. Xu,et al., Eur. J. Med. Chem. 217 (2021) 113380. DOI:10.1016/j.ejmech.2021.113380 |
| [33] |
S. Xu,L. Sun,B. Huang,et al., Future Med. Chem. 12 (2020) 1281-1284. DOI:10.4155/fmc-2020-0084 |
| [34] |
S. Fox,S. Farr-Jones,L. Sopchak,A. Boggs,J. Comley, SLAS Discov. 9 (2004) 354-358. DOI:10.1177/1087057104265290 |
| [35] |
T. Pan,Y. Ding,L. Wu,et al., Eur. J. Med. Chem. 166 (2019) 480-501. DOI:10.1016/j.ejmech.2019.01.059 |
| [36] |
A.D. Huber,D.L. Pineda,D. Liu,et al., ACS Infect. Dis. 5 (2019) 750-758. DOI:10.1021/acsinfecdis.8b00235 |
| [37] |
X. Zhang,J. Cheng,J. Ma,et al., ACS Infect. Dis. 5 (2019) 759-768. DOI:10.1021/acsinfecdis.8b00269 |
| [38] |
Y. Pei,C. Wang,H. Ben,et al., ACS Infect. Dis. 5 (2019) 778-787. DOI:10.1021/acsinfecdis.9b00030 |
| [39] |
M. Yamasaki,N. Matsuda,K. Matoba,et al., Virus Res. 306 (2021) 198565. DOI:10.1016/j.virusres.2021.198565 |
| [40] |
O. Slater,M. Kontoyianni, Expert Opin. Drug Discov. 14 (2019) 619-637. DOI:10.1080/17460441.2019.1604677 |
| [41] |
M. Toyama,N. Sakakibara,M. Takeda,et al., Virus Res. 271 (2019) 197677. DOI:10.1016/j.virusres.2019.197677 |
| [42] |
S. Senaweera,H. Du,H. Zhang,et al., Viruses 13 (2021) 770. DOI:10.3390/v13050770 |
| [43] |
Y. Wang,Z. Wang,J. Liu,et al., Bioorg. Med. Chem. 36 (2021) 116096. DOI:10.1016/j.bmc.2021.116096 |
| [44] |
Y. Yang,Y. Yan,J. Yin,et al., Viruses 14 (2022) 348. DOI:10.3390/v14020348 |
| [45] |
D. Yang,L. Wang,P. Yuan,et al., Chin. Chem. Lett. 34 (2023) 107964. DOI:10.1016/j.cclet.2022.107964 |
| [46] |
J. Zhu,C. Chen,J. Dong,et al., Chin. Chem. Lett. 34 (2023) 107514. DOI:10.1016/j.cclet.2022.05.028 |
| [47] |
S. Gao,T. Huang,L. Song,et al., Acta Pharm. Sin. B 12 (2022) 581-599. DOI:10.1016/j.apsb.2021.08.027 |
| [48] |
S. Pushpakom,F. Iorio,P.A. Eyers,et al., Nat. Rev. Drug Discov. 18 (2019) 41-58. DOI:10.1038/nrd.2018.168 |
| [49] |
P. Zhan,B. Yu,L. Ouyang, Drug Discov. Today 27 (2022) 1785-1788. DOI:10.1016/j.drudis.2022.05.026 |
| [50] |
X.F. Wei,C.Y. Gan,J. Cui,et al., Antimicrob. Agents Chemother. 62 (2018) e01302-18. |
| [51] |
J. Blaising,S.J. Polyak,E.I. Pécheur, Antiviral Res. 107 (2014) 84-94. DOI:10.1016/j.antiviral.2014.04.006 |
| [52] |
J.A. Kang,S. Kim,M. Park,et al., Nat. Commun. 10 (2019) 2184. DOI:10.1038/s41467-019-10200-5 |
| [53] |
H.W. Seo,J.P. Seo,Y. Cho,et al., Virus Res. 263 (2019) 102-111. DOI:10.1016/j.virusres.2019.01.004 |
| [54] |
X. Mao,D.L. Auer,W. Buchalla,et al., Agents Chemother. 64 (2020) e00576-20. |
| [55] |
Y. Xiao,C. Liu,W. Tang,H. Zhang,X. Chen, Front. Microbiol. 10 (2019) 2638. DOI:10.3389/fmicb.2019.02638 |
| [56] |
D.L. Ma,D.S.H. Chan,C.H. Leung, Chem. Soc. Rev. 42 (2013) 2130-2141. DOI:10.1039/c2cs35357a |
| [57] |
J. Du,J. Guo,D. Kang,et al., Chin. Chem. Lett. 31 (2020) 1695-1708. DOI:10.1016/j.cclet.2020.03.028 |
| [58] |
J.W.H. Li,J.C. Vederas, Science 325 (2009) 161-165. DOI:10.1126/science.1168243 |
| [59] |
Z. Guo, Acta Pharm. Sin. B 7 (2017) 119-136. DOI:10.3390/sym9070119 |
| [60] |
L. Zhao,X. Xiong,L. Liu,et al., Chin. Chem. Lett. 33 (2022) 1841-1849. DOI:10.3390/biology11121841 |
| [61] |
H.J. Chen,W.L. Wang,G.F. Wang,et al., ChemMedChem 3 (2008) 1316-1321. DOI:10.1002/cmdc.200800136 |
| [62] |
L. Yang,L. Shi,H. Chen,et al., Acta Pharmacol. Sin. 35 (2014) 410-418. DOI:10.1038/aps.2013.175 |
| [63] |
M. Luo,Z. Chen,M. Liu,et al., J. Med. Virol. 94 (2022) 2727-2735. DOI:10.1002/jmv.27621 |
| [64] |
H. Ju,J. Zhang,B. Huang,et al., J. Med. Chem. 60 (2017) 3533-3551. DOI:10.1021/acs.jmedchem.6b01227 |
| [65] |
Y. Yao,Z. Liu,M. Zhao,et al., Acta Pharm. Sin. B 10 (2020) 1453-1475. DOI:10.1016/j.apsb.2020.04.002 |
| [66] |
Y. Tao,X. Hao,X. Ding,et al., Eur. J. Med. Chem. 201 (2020) 112374. DOI:10.1016/j.ejmech.2020.112374 |
| [67] |
L. Li,A. Wang,B. Wang,et al., Chin. Chem. Lett. 31 (2020) 409-412. DOI:10.1016/j.cclet.2019.07.038 |
| [68] |
K. Vandyck,G. Rombouts,B. Stoops,et al., J. Med. Chem. 61 (2018) 6247-6260. DOI:10.1021/acs.jmedchem.8b00654 |
| [69] |
Q. Ren,X. Liu,G. Yan,et al., J. Med. Chem. 61 (2018) 1355-1374. DOI:10.1021/acs.jmedchem.7b01914 |
| [70] |
J. Tang,A.D. Huber,D.L. Pineda,et al., Eur. J. Med. Chem. 164 (2019) 179-192. DOI:10.1016/j.ejmech.2018.12.047 |
| [71] |
H.G. Na,A. Imran,K. Kim,et al., ACS Med. Chem. Lett. 11 (2020) 166-171. DOI:10.1021/acsmedchemlett.9b00550 |
| [72] |
Y.H. Lee,H.M. Cha,J.Y. Hwang,et al., ACS Med. Chem. Lett. 12 (2021) 242-248. DOI:10.1021/acsmedchemlett.0c00606 |
| [73] |
W. Kim,J.A. Kang,M. Park,et al., J. Med. Chem. 64 (2021) 5500-5518. DOI:10.1021/acs.jmedchem.0c01938 |
| [74] |
K. Spunde,B. Vigante,U.N. Dubova,et al., Pharmaceuticals 15 (2022) 773. DOI:10.3390/ph15070773 |
| [75] |
K. Lv,S. Wu,Z. Tao,et al., Eur. J. Med. Chem. 228 (2022) 113974. DOI:10.1016/j.ejmech.2021.113974 |
| [76] |
S.D. Kuduk,L.G. DeRatt,B. Stoops,et al., Bioorg. Med. Chem. Lett. 72 (2022) 128823. DOI:10.1016/j.bmcl.2022.128823 |
| [77] |
S.D. Kuduk,B. Stoops,A.M. Lam,et al., Bioorg. Med. Chem. Lett. 52 (2021) 128353. DOI:10.1016/j.bmcl.2021.128353 |
| [78] |
D. Wu,X. Zheng,R. Liu,et al., Acta Pharm. Sin. B 12 (2022) 1351-1362. DOI:10.1016/j.apsb.2021.09.027 |
| [79] |
Y. Yang,S. Zhang,Q. Zhou,et al., Acta Pharm. Sin. B 10 (2020) 2339-2347. DOI:10.1016/j.apsb.2020.04.003 |
| [80] |
L. Wang,M.C. Casey,S.K.V. Vernekar,et al., Acta Pharm. Sin. B 11 (2021) 810-822. DOI:10.1016/j.apsb.2020.07.016 |
| [81] |
D. Feng,F. Wei,Y. Sun,et al., Chin. Chem. Lett. 32 (2021) 4053-4057. DOI:10.1016/j.cclet.2021.02.033 |
| [82] |
K. Lv,S. Wu,W. Li,et al., Bioorg. Chem. 94 (2020) 103363. DOI:10.1016/j.bioorg.2019.103363 |
| [83] |
W. Chen,F. Liu,Q. Zhao,et al., J. Med. Chem. 63 (2020) 8134-8145. DOI:10.1021/acs.jmedchem.0c00346 |
| [84] |
F. Amblard,S. Boucle,L. Bassit,et al., Bioorg. Med. Chem. 31 (2021) 115952. DOI:10.1016/j.bmc.2020.115952 |
| [85] |
S.J. Hurwitz,N. McBrearty,A. Arzumanyan,et al., Viruses 13 (2021) 114. DOI:10.3390/v13010114 |
| [86] |
Y. Ma,S. Zhao,Y. Ren,et al., Eur. J. Med. Chem. 225 (2021) 113780. DOI:10.1016/j.ejmech.2021.113780 |
| [87] |
A.G. Cole,S.G. Kultgen,N. Mani,et al., RSC Med. Chem. 13 (2022) 343-349. DOI:10.1039/d1md00318f |
| [88] |
C. Viegas-Junior,A. Danuello,V. da Silva Bolzani,E.J. Barreiro,C.A. Fraga, Curr. Med. Chem. 14 (2007) 1829-1852. DOI:10.2174/092986707781058805 |
| [89] |
H. Jia,J. Yu,X. Du,et al., Eur. J. Med. Chem. 202 (2020) 112495. DOI:10.1016/j.ejmech.2020.112495 |
| [90] |
X. Li,Z. Zhang,Y. Chen,et al., ACS Med. Chem. Lett. 13 (2022) 507-512. DOI:10.1021/acsmedchemlett.2c00002 |
| [91] |
L. Liu,M. Wang,C. Li,et al., Bioorg. Chem. 128 (2022) 106052. DOI:10.1016/j.bioorg.2022.106052 |
| [92] |
S. Wang,Y. Ren,Q. Li,et al., Bioorg. Chem. 129 (2022) 106192. DOI:10.1016/j.bioorg.2022.106192 |
| [93] |
S. Zhang,J. Zhang,P. Gao,et al., Drug Discov. Today 24 (2019) 805-813. DOI:10.1016/j.drudis.2018.11.021 |
| [94] |
H. Guo,X. Zhuang,K. Qian,et al., Acta Pharm. Sin. B 2 (2012) 213-219. DOI:10.1016/j.apsb.2012.02.008 |
| [95] |
J.B. Zawilska,J. Wojcieszak,A.B. Olejniczak, Pharmacol. Rep. 65 (2013) 1-14. DOI:10.1016/s1734-1140(13)70959-9 |
| [96] |
X. Ji,X. Jiang,C. Kobayashi,et al., Molecules 27 (2022) 5987. DOI:10.3390/molecules27185987 |
| [97] |
Z. Fang,Y. Song,P. Zhan,Q. Zhang,X. Liu, Future Med. Chem. 6 (2014) 885-901. DOI:10.4155/fmc.14.50 |
| [98] |
K. Tang,S. Wang,W. Gao,Y. Song,B. Yu, Acta Pharm. Sin. B 12 (2022) 4309-4326. DOI:10.1016/j.apsb.2022.09.022 |
| [99] |
C. Wang,Y. Pei,L. Wang,et al., J. Med. Chem. 63 (2020) 6066-6089. DOI:10.1021/acs.jmedchem.0c00292 |
| [100] |
B. Huang,D. Kang,P. Zhan,X. Liu, Expert Opin. Drug Discov. 10 (2015) 1271-1281. DOI:10.1517/17460441.2015.1083007 |
| [101] |
Q. Nie,S. Zhong,Y. Li,G. Zhang,Y. Li, Chin. Chem. Lett. 33 (2022) 2559-2563. DOI:10.1016/j.cclet.2021.09.041 |
| [102] |
K. Klumpp,T. Crépin, Curr. Opin. Virol. 5 (2014) 63-71. DOI:10.1016/j.coviro.2014.02.002 |
| [103] |
A.C.H. Lee,E.P. Thi,A. Ardzinski,et al., J. Hepatol. 73 (2020) S833. |
| [104] |
C. Abadzapatero,J. Metz, Drug Discov. Today 10 (2005) 464-469. DOI:10.1016/S1359-6446(05)03386-6 |
| [105] |
P.D. Leeson,B. Springthorpe, Nat. Rev. Drug Discov. 6 (2007) 881-890. DOI:10.1038/nrd2445 |
| [106] |
X. Jiang,J. Yu,Z. Zhou,et al., Med. Res. Rev. 39 (2019) 2194-2238. DOI:10.1002/med.21581 |
| [107] |
W. Wei,S. Cherukupalli,L. Jing,X. Liu,P. Zhan, Drug Discov. Today 25 (2020) 1839-1845. DOI:10.1016/j.drudis.2020.07.017 |
| [108] |
Y. Wang,X. Jiang,F. Feng,W. Liu,H. Sun, Acta Pharm. Sin. B 10 (2020) 207-238. DOI:10.3390/molecules25010207 |
| [109] |
M. de Wispelaere,G. Du,K.A. Donovan,et al., Nat. Commun. 10 (2019) 3468. DOI:10.1038/s41467-019-11429-w |
| [110] |
H. Wu,H. Yao,C. He,et al., Acta Pharm. Sin. B 12 (2022) 3548-3566. DOI:10.1016/j.apsb.2022.03.019 |
| [111] |
S. De Cesco,J. Kurian,C. Dufresne,et al., Eur. J. Med. Chem. 138 (2017) 96-114. DOI:10.1016/j.ejmech.2017.06.019 |