 2023, Vol. 34
2023, Vol. 34 


b Key Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China;
c Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
The widespread use of unsustainable and nondegradable petroleum-based plastics, such as polypropylene, polyethylene, and polystyrene, has brought serious environmental crisis [1–3]. To solve this issue, design and synthesis of green alternatives for petroleum-based materials that can be recycled to its initial monomer or value-added chemicals becomes a crucial topic [4–9]. It is worth noting that several significant advances have been made in this emerging field (Scheme 1) [10–37]. For example, in 2015, for the traditionally "non-polymerizable" monomer γ-butyrolactone, ring-opening polymerization (ROP) at low temperature was successfully achieved by Chen and Hong [12]. Due to the inherently low ceiling temperature, it was easily depolymerized back to the initial monomer. Afterwards, to address material depolymerizability and performance trade-off, an ingenious strategy of introducing fused-ring was implemented by the same group [13]. The bicyclic fused γ-butyrolactone monomer 3,4-T6GBL could be polymerized steadily at room temperature. Meanwhile, the obtained polyester exhibited good mechanical properties, while maintaining high degradability. In 2021, Tao prepared a series of dithiolactone monomers derived from abundant feedstock α-amino acids. Noting that the resulting polythioesters after ROP exhibited high crystallinity, structural diversity as well as outstanding chemical recyclability [30]. Ring-closing depolymerizations could be achieved in dilute solution at 25 ℃ with quantitative yields of recovered monomers. Recently, a kind of novel polyolefin-like aromatic polyester materials with functionalizable molecules were synthesized by Zhu. While heating to 120 ℃, these materials could be thermally recycled back to their original monomers [32].

|
Download:
|
| Scheme 1. The polymerization-depolymerization cycle of representative monomers. | |
Despite the above breakthroughs, none of these cases is of practical value. Currently, only few polyesters, such as poly(lactide) (PLA), poly(ε-caprolactone) (PCL) and polyhydroxyalkanoates (PHA), are gaining practical applications [38,39]. Hence, the recycling of these kinds of polymers should also pay significant attention. For example, PLLA, derived from lactic acid, is already commercially available and widely used in packaging, textile and biomedical application areas [40–44]. While, biodegradation process of PLLA occurs only under certain environmental conditions [45]. If not handled properly, PLLA still might be a source of environmental pollution. On the other hand, chemical recycling of end-of-life PLA plastics has many benefits. First, it helps to mitigate possible environmental pollution and achieve the secondary utilization of waste resources. Second, it improves the recycling efficiency of PLA plastics. Third, it saves a lot of land, energy and time to reproduce lactic acid. Therefore, it is very significant to achieve the chemical recycling of PLA.
Considerable effort has been exerted on chemical recycling of PLA in recent years [46]. However, chemical recycling of PLA polymer to recover L-LA monomer remains a key challenge. Typically, in the process of high temperature thermal degradation, a variety of degradation products are produced, such as LA diastereomers (L-LA, D-LA and meso–LA), cyclic oligomers, CO2, CO, CH3CHO and CH2=CHCOOH [47,48]. The recovery of lactide with high selectivity is of great significance [49]. On the other hand, the alcoholysis is an alternative strategy to transform PLA into high value-added lactate ester which can be used as an environmentally friendly solvent [50]. Alcoholysis of PLA has been studied extensively [51–60], but there are still some problems that have not been well solved, such as highly efficient depolymerization under mild conditions. Therefore, there exists great development space and vast prospective in chemical recycling of PLLA.
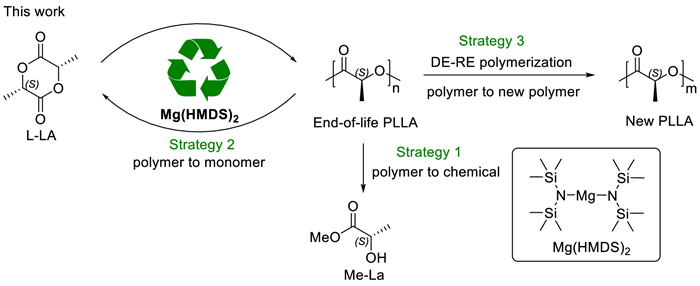
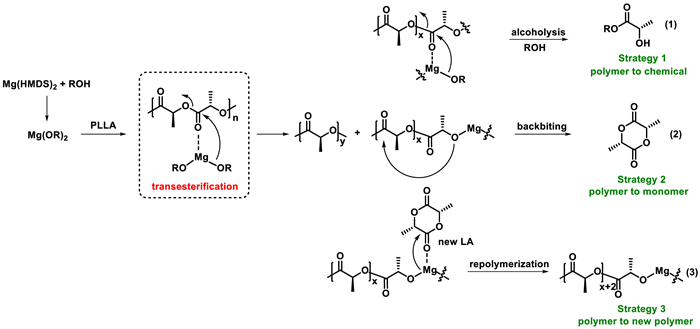
Our research group is committed to seeking some multifunctional catalysts in polymer synthesis and degradation [54]. The characteristics of abundant, biocompatible and environmentally benign make magnesium-based catalysts an attractive catalyst. In our polymerization exploration of magnesium bis[bis(trimethylsilyl)amide] [Mg(HMDS)2], the presence of transesterification and chain transfer were revealed. Due to rapid and reversible chain transfer, the polymerizations exhibited controllable as well as immortal features. Taking advantage of transesterification, we investigated three different strategies to convert end-of-life poly(lactide) into lactate ester, lactide, and new poly(lactide) using Mg(HMDS)2 as a promoter (Scheme 2). The first strategy was to transform poly(lactide) into high value-added lactate ester by alcoholysis under mild conditions. The second strategy was the most direct "polymerization-depolymerization" cycle. In this part, closed-loop recycling from polylactide to lactide monomer was achieved. In addition, based on random depolymerization and controllable polymerization, a new "depolymerization-repolymerization" strategy was explored to transform poly(lactide) into new poly(lactide). This work provides an environmentally benign, versatile catalyst system for polymer synthesis and degradation. These degradation strategies greatly increase the recycling efficiency of the poly(lactide) materials.

|
Download:
|
| Scheme 2. Three pathways to recycle poly(lactide). | |
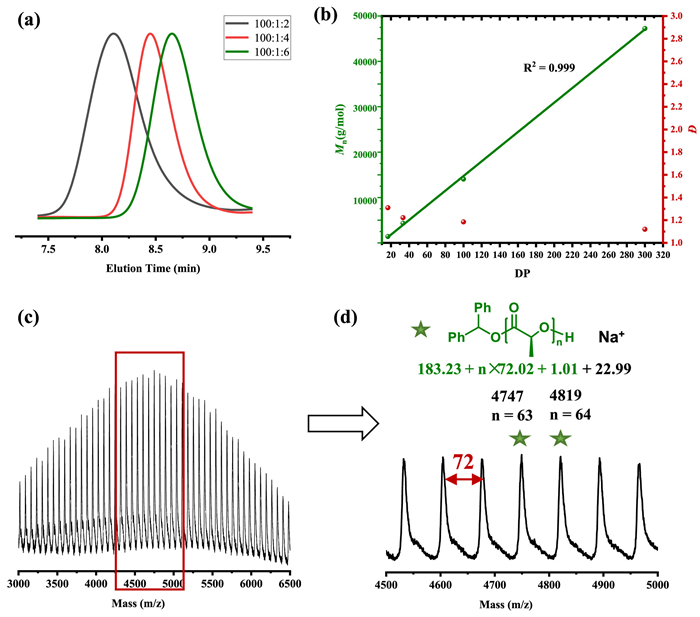
The catalytic performance of Mg(HMDS)2 in the ROP of L-LA was first investigated in DCM at room temperature. The ROP results were summarized in Table 1. When Mg(HMDS)2 was employed alone to initiate the polymerization, the reaction proceeded slowly and only 10% monomer conversion could be detected after 3 h, demonstrating the Mg-N species initiation was inefficient (entry 1). It has been reported that metal alkoxide generally shows better initiation efficiency and controllability in ROP than its amide analogues [12]. The 1H NMR spectrum showed that when Mg(HMDS)2 and alcohol were mixed, HN(SiMe3)2 appeared and the magnesium alkoxide species formed (Fig. S1 in Supporting information). Therefore, in situ alcoholysis of benzyl alcohol (BnOH) with Mg(HMDS)2 was manipulated to generate the Mg alkoxide species. When the [Cat.]0/[OH]0 ratio was 1/1, the molecular weight of the produced PLA was much higher than that of theoretical molecular weight (Table S1 in Supporting information, 14.0 kg/mol versus 27.4 kg/mol), coupled with broad dispersity (Đ = 1.63). Increasing the amount of alcohol to 2 equivalents, the ROP process became more controllable. After 8 h of reaction, almost complete conversion was achieved and the molecular weight was consistent with the theoretical value (entry 2, 7.1 kg/mol versus 8.9 kg/mol), accompanied by a narrow dispersity (Đ = 1.22). This result illustrated that 2 equiv. of alcohol was necessary to completely replace the amino group and magnesium alkoxide was the real active species. Previous studies suggested that alcohol and metal may form clusters thereby affecting the reaction [61], for this reason, diphenyl methanol with large steric hindrance was taken into account. Surprisingly, an excellent polymerization activity was highlighted by the fact that 99% conversion could be realized within 1 min (entry 3). This result indicting that the aggregation of the metal may be broken in this condition thus promoting the interaction of the monomer with the metal center. Moreover, when the amount of alcohol was further increased to 4 or 6 equivalents, the molecular weights were found to be closer to the theoretical values (entry 4, 3.7 kg/mol versus 3.9 kg/mol; entry 5, 2.5 kg/mol versus 2.5 kg/mol), along with a decrease in dispersity (Fig. 1a, 1.43 versus 1.24). The phenomenon proved that Mg(HMDS)2 could mediate the immortal ROP, benefited from the chain transfer, which was the rapid and reversible exchange reaction between the active species and alcohol [62]. Besides, increasing the ratio of [L-LA]0/[I]0 from 100/6 to 200/6, 600/6 and 2000/6, all the polymerizations performed efficiently (entries 6–8), the molecular weight had a linear relationship with the degree of polymerization (Fig. 1b), exhibiting the characteristics of controllable and living polymerization [63]. In addition, the backbone structure of the polymer was analyzed by MALDI-TOF mass spectroscopy. As shown in Figs. 1c and d, the poly(lactide) was composed of the Ph2CHOH moiety at the initiating terminal and an intact hydroxyl end-group at the capping terminal. High chain end fidelity confirmed that polymerization was indeed initiated by Ph2CHOH. However, it was worth noting that the interval between two adjacent m/z peaks was 72, which indicated that a transesterification reaction occurred [64]. Due to the rapid chain transfer, the adverse effect of transesterification on polymerization was inhibited.
|
|
Table 1 Results for ROP of L-LA catalyzed by Mg(HMDS)2. |
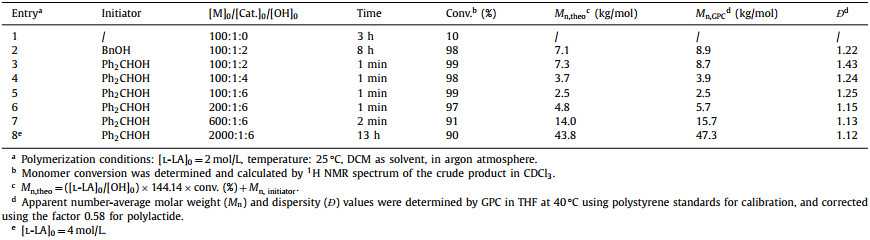

|
Download:
|
| Fig. 1. (a) GPC analysis of ROP of L-LA at different DPs (Table 1, entries 3–5). (b) Plot of DP ([M]0/[I]0) versus Mn and Đ (Table 1, entries 5–8, R2 = 0.999). (c, d) MALDI-TOF analysis of the obtained PLLA catalyzed by Mg(HMDS)2. | |
After the discovery of Mg(HMDS)2/alcohol catalyzed transesterification, a supposition was raised whether transesterification could be used in the degradation process of PLA. Taking advantage of transesterification, the Mg alkoxide species could coordinate with PLA to initiate chain scission. Then, three possible routes to achieve the transformation of poly(lactide) were proposed as shown in Scheme 3. For the first pathway, Mg alkoxide species further attacks the carbonyl group, if there is enough alcohol, the polymer chain eventually degrades to lactate ester. For the second pathway, Mg alkoxide further attacks the vicinal carbonyl group via a backbiting reaction and the polymer chain then depolymerizes into the corresponding monomer lactide. In the third route, adding new monomer (lactide) to the reaction system, Mg alkoxide might undergo further polymerization to produce a new polymer chain. In order to verify the possibility of three strategies, the corresponding experiments were conducted next.

|
Download:
|
| Scheme 3. Possible degradation pathway of PLLA. | |
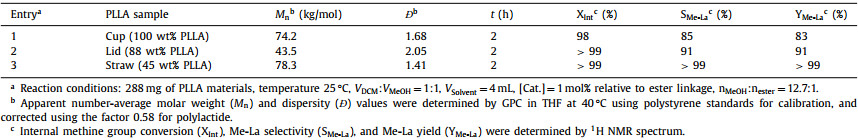
First, transformation of poly(lactide) to high value-added lactate ester by alcoholysis was performed. The alcoholysis reactions were performed using 1 mol% catalyst with respect to the ester linkages of PLLA, and MeOH as alcoholysis reagent. The PLLA commodities with different PLLA contents were purchased, such as cup (100 wt% PLLA), lid (88 wt% PLLA), and straw (45 wt% PLLA) (Table 2, entries 1–3). The molecular weights of all materials were characterized by GPC (Figs. S12-S14 in Supporting information). The polymer materials were sheared into fragments and reacted directly without other treatments. To our delight, depolymerizations of all three commodities efficiently proceeded at room temperature within 2 h, yielding the corresponding methyl lactate (Me-La) in high yields (83%−99%) and selectivities (85%−99%). These results showed that the purity and molecular weight of the PLLA commodities have few effects on the alcoholysis reaction, which also manifested the robustness and efficiency of this catalyst.
|
|
Table 2 Methanolysis of different PLLA materials catalyzed by Mg(HMDS)2. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
To clarify the mechanism of the depolymerization process, concentration changes of different components during depolymerization was investigated. According to the integration of different methines (Fig. S2 in Supporting information), internal, chain end, Me-La), their contents were calculated respectively. It could be observed that at the beginning of the reaction, the internal methine groups rapidly converted to chain end methine, indicating that the polymer underwent rapid fracture. After that, methyl lactate was gradually formed (Fig. S3 in Supporting information). This phenomenon implied that the depolymerization process was based on the mechanism of random degradation rather than unzipping degradation.
Encouraged by the above result, a gram-scale reaction was carried out. When 16.32 g commercial PLLA materials were used, depolymerization process could still be achieved at room temperature within 2 h. After simple filtration and concentration, 20.84 g of pure Me-La was separated with a yield of 90% (Fig. 2 and Fig. S6 in Supporting information). In addition, the optical purity of methyl lactate obtained from depolymerization of PLLA was detected by HPLC analysis. The content of L-configuration methyl lactate was demonstrated as 99% (Fig. S5 in Supporting information). The high optical purity proved that the racemization reaction during depolymerization could be ignored. The above experimental results showed that depolymerization strategy 1 promoted by Mg(HMDS)2 has been successfully implemented.

|
Download:
|
| Fig. 2. Depolymerization of PLLA materials catalyzed by Mg(HMDS)2 (Reaction conditions: 16.32 g of PLLA materials, room temperature, VDCM: VMeOH = 1:1, VSolvent = 226 mL, [Cat.] = 1 mol% relative to ester linkage, nMeOH: nester = 12.7:1. The yield of Me-La was 90%). | |
{kind=link}
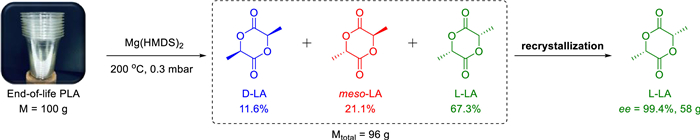
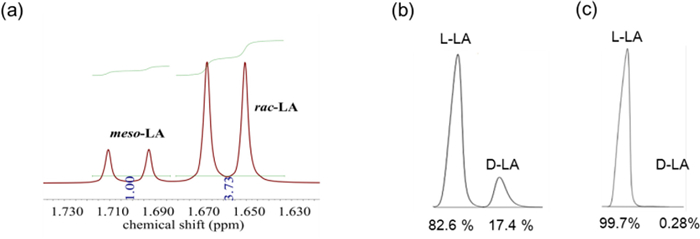
As far as we known, the ring-opening polymerization of LA is enthalpy favourable (ΔH < 0) and entropy unfavorable (ΔS < 0). Based on the formula ΔG = ΔH - TΔS, there is a ceiling temperature (Tc) at which ΔG is equal to zero and polymerization-depolymerization is at equilibrium. When the temperature is higher than Tc, the depolymerization of polymer is favorable [65]. However, for LA, a high Tc (in bulk) of 914 K was reported [66]. While, we believe that timely removal of LA from the depolymerization system by distillation will disturb the equilibrium of the reaction and promote the depolymerization. Therefore, the reaction conditions of high temperature and low pressure are suitable. On the other hand, the addition of metal catalysts is necessary to promote the backbiting reaction of intramolecular transesterification. Chemical recycling of the high molecular weight PLLA, especially after-use PLLA goods, to recover L-LA remains a significant challenge. Hence, there is a strong drive for us to explore the chemical recycling of high molecular weight end-of-life PLLA plastic waste back to lactide monomer by using Mg(HMDS)2 (Scheme 4). Strikingly, employing 100 g PLLA cup with molecular weight of 74.2 kg/mol as raw material, the depolymerization process catalyzed by Mg(HMDS)2 was smoothly achieved under 200 ℃ and 0.3 mbar conditions, resulting in 96 g crude lactide. Even the crude product was detected with high purity by 1H NMR (Fig. S7 in Supporting information), Moreover, the high yield further indicated that there were almost no other side reactions in this process. The proportions of L-LA, D-LA and meso–LA in the crude product were 67.3%, 11.6%, 21.1%, respectively, which was confirmed by 1H NMR spectrum and HPLC analysis (Figs. 3a and b). Similar to making L-LA from PLLA oligomers in industrial production, purification processes are needed [67]; [68]. The meso–LA products were completely removed and the enantiomeric excess (ee) value of L-LA reached > 99% after five times of recrystallization in toluene as shown in the HPLC analysis (Fig. 3c, Figs. S8 and S9 in Supporting information). In addition, the above purified L-LA (Scheme 4) was also performed the ROP process at room temperature (Table S3 in Supporting information). These above experiments confirmed that Mg(HMDS)2 can realize the "polymerization-depolymerization" cycle of L-LA (strategy 2). As expected, this polymerization was implemented controllable as well. This is one of the few examples where a catalyst can simultaneously achieve "polymerization-depolymerization" cycle.

|
Download:
|
| Scheme 4. The depolymerization to recover L-LA. Depolymerization conditions: 100 g of PLLA cup, 5.6 mmol Mg(HMDS)2, 200 ℃, 0.3 mbar. | |
{kind=link}

|
Download:
|
| Fig. 3. (a) 1H NMR spectrum of crude lactide (meso–LA: rac-LA = 21.1:78.9) (400 MHz, CDCl3, 298 K). (b) HPLC analysis of crude lactide (L-LA: D-LA = 82.6:17.4). (c) HPLC analysis of purified lactide (L-LA: D-LA = 99.7:0.3). | |
{kind=link}
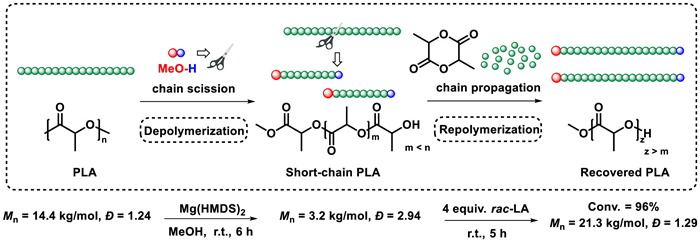
At last, these unique characteristics of Mg(HMDS)2 stimulate us to develop an approach to maximize both its depolymerization and polymerization abilities. Therefore, a new "depolymerization-repolymerization" strategy was proposed. First, PLA polymer was depolymerized into short-chain polymers, utilizing a Mg(HMDS)2 catalyzed random methanolysis process. Then, the short-chain polymers were repolymerized with newly added lactide to form new PLA materials. As shown in Fig. 4, using 144 mg PLA sample synthesized in our laboratory as the initial polymer, 48 µmol MeOH (4 equiv. relative to polymer chain) was added as chain scission reagent to start the depolymerization process. After 6 h, the molecular weight of the polymer was reduced to 3.2 kg/mol (a quarter of its original value), which means that the transformation process of polymer to oligomers occurred (Fig. S11 in Supporting information). The broad dispersity further illustrated that this methanolysis process was random. Afterwards, 4 equiv. rac-LA was added to the reaction mixture, the repolymerization was successfully begun, yielding the final PLLA with a molecular weight of 21.3 kg/mol and a dispersity of 1.29 (Fig. S10 in Supporting information). Interestingly, a narrowing of dispersity upon repolymerization could be observed, suggesting that short-chain polymers propagated faster than long-chain polymers [69]. Despite this "polymer to new polymer" strategy did not achieve complete closed-loop chemical recycling, it demonstrates a new concept to plastics sustainability and maximizes the recovery efficiency of PLA.

|
Download:
|
| Fig. 4. DE-RE polymerization of PLA. DE-polymerization conditions: 144 mg of PLA (12 µmol polymer chain), 48 µmol MeOH (4 equiv. relative to polymer chain), 24 µmol Mg(HMDS)2, room temperature. RE-polymerization conditions: 4 equiv. rac-LA and 4 mL DCM were added, room temperature. | |
{kind=link}
In summary, Mg(HMDS)2 has successfully promoted the ring-opening polymerization of lactide and chemical upgrading of end-of-life poly(lactide) plastics to lactide (polymer to monomer), lactate ester (polymer to chemical) and new poly(lactide) (polymer to new polymer). Mg(HMDS)2 represents a rare example which can achieve a variety of depolymerization and polymerization strategies. Using Mg(HMDS)2/Ph2CHOH as catalytic system, the effective polymerization of lactide was carried out and a well-defined poly(lactide) was obtained. Polymerizations were characterized by being controllable and immortal. Taking advantage of transesterification discovered during the reaction, the efficient alcoholysis of 16.3 g commercial PLLA materials was achieved at room temperature, and 20.8 g of pure methyl lactate was gained after filtration and concentration. On the other hand, depolymerization of 100 g PLLA cup catalyzed by Mg(HMDS)2 was smoothly achieved under 200 ℃ and 0.3 mbar conditions, resulting in 96 g crude lactide. The purity of L-LA reached > 99% after recrystallization. Moreover, based on the simultaneous realization of random depolymerization and controlled polymerization, a new "depolymerization-repolymerization" strategy was also carried out to directly convert poly(lactide) into new poly(lactide) under mild conditions. Despite this "polymer to new polymer" strategy did not achieve complete closed-loop chemical recycling, it represents a promising concept that can be deeply explored. These three strategies greatly improved the recycling efficiency of poly(lactide). Further investigations of this new strategy are in progress in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was support by the National Natural Science Foundation of China (No. 21901249), Taishan Scholars Program of Shandong Province (No. tsqn201812112) and the Scientific Research and Innovation Fund Project of Shandong Energy Research Institute (No. SEI I202004).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108158.
| [1] |
M. MacLeod, H.P.H. Arp, M.B. Tekman, A. Jahnke, Science 373 (2021) 61-65. DOI:10.1126/science.abg5433 |
| [2] |
R.A. Sheldon, M. Norton, Green Chem. 22 (2020) 6310-6322. DOI:10.1039/D0GC02630A |
| [3] |
J.C. Worch, A.P. Dove, ACS Macro Lett. 9 (2020) 1494-1506. DOI:10.1021/acsmacrolett.0c00582 |
| [4] |
D.K. Schneiderman, M.A. Hillmyer, Macromolecules 50 (2017) 3733-3749. DOI:10.1021/acs.macromol.7b00293 |
| [5] |
X. Tang, E.Y.X. Chen, Chem 5 (2019) 284-312. DOI:10.1016/j.chempr.2018.10.011 |
| [6] |
G.W. Coates, Y.D.Y.L. Getzler, Nat. Rev. Mater. 5 (2020) 501-516. DOI:10.1038/s41578-020-0190-4 |
| [7] |
X.B. Lu, Y. Liu, H. Zhou, Chem. Eur. J. 24 (2018) 11255-11266. DOI:10.1002/chem.201704461 |
| [8] |
J.M. Garcia, M.L. Robertson, Science 358 (2017) 870-872. DOI:10.1126/science.aaq0324 |
| [9] |
G. Xu, Q. Wang, Green Chem 24 (2022) 2321-2346. DOI:10.1039/D1GC03901F |
| [10] |
A. Rahimi, J.M. García, Nat. Rev. Chem. 1 (2017) 0046. DOI:10.1038/s41570-017-0046 |
| [11] |
Y. Zhu, C. Romain, C.K. Williams, Nature 540 (2016) 354-362. DOI:10.1038/nature21001 |
| [12] |
M. Hong, E.Y.X. Chen, Nat. Chem. 8 (2016) 42-49. DOI:10.1038/nchem.2391 |
| [13] |
J.B. Zhu, E.M. Watson, J. Tang, E.Y.X. Chen, Science 360 (2018) 398-403. DOI:10.1126/science.aar5498 |
| [14] |
B.A. Abel, R.L. Snyde, G.W. Coates, Science 373 (2021) 783-789. DOI:10.1126/science.abh0626 |
| [15] |
M. Li, Y. Tao, J. Tang, et al., J. Am. Chem. Soc. 141 (2019) 281-289. DOI:10.1021/jacs.8b09739 |
| [16] |
J. Lian, M. Li, S. Wang, Y. Tao, X. Wang, Macromolecules 53 (2020) 10830-10836. DOI:10.1021/acs.macromol.0c02065 |
| [17] |
J. Chen, Y. Dong, C. Xiao, Y. Tao, X. Wang, Macromolecules 54 (2021) 2226-2231. DOI:10.1021/acs.macromol.0c02689 |
| [18] |
M. Haussler, M. Eck, D. Rothauer, S. Mecking, Nature 590 (2021) 423-427. DOI:10.1038/s41586-020-03149-9 |
| [19] |
R. Yang, G. Xu, B. Dong, X. Guo, Q. Wang, ACS Sustain. Chem. Eng. 10 (2022) 9860-9871. DOI:10.1021/acssuschemeng.2c01708 |
| [20] |
M. Hong, E.Y.X. Chen, Angew. Chem. Int. Ed. 55 (2016) 4188-4193. DOI:10.1002/anie.201601092 |
| [21] |
J. Su, G. Xu, B. Dong, et al., Polym. Chem. 13 (2022) 5897-5904. DOI:10.1039/D2PY00953F |
| [22] |
L.G. Li, Q.Y. Wang, Q.Y. Zheng, F.S. Du, Z.C. Li, Macromolecules 54 (2021) 6745-6752. DOI:10.1021/acs.macromol.1c00497 |
| [23] |
Y. Liu, H. Zhou, J.Z. Guo, W.M. Ren, X.B. Lu, Angew. Chem. Int. Ed. 56 (2017) 4862-4866. DOI:10.1002/anie.201701438 |
| [24] |
J.P. MacDonald, M.P. Shaver, Polym. Chem. 7 (2016) 553-559. DOI:10.1039/C5PY01606A |
| [25] |
M.M. Beromi, C.R. Kennedy, J.M. Younker, et al., Nat. Chem. 13 (2021) 156-162. DOI:10.1038/s41557-020-00614-w |
| [26] |
B. Dong, G. Xu, R. Yang, Q. Wang, Chem. Asian J. 17 (2022) e202200667. DOI:10.1002/asia.202200667 |
| [27] |
D. Sathe, J. Zhou, H. Chen, et al., Nat. Chem. 13 (2021) 743-750. DOI:10.1038/s41557-021-00748-5 |
| [28] |
Y. Shen, W. Xiong, Y. Li, et al., CCS Chem. 3 (2021) 620-630. DOI:10.31635/ccschem.020.202000232 |
| [29] |
C. Shi, Z.C. Li, L. Caporaso, et al., Chemistry 7 (2021) 670-685. DOI:10.1016/j.chempr.2021.02.003 |
| [30] |
Y. Wang, M. Li, J. Chen, Y. Tao, X. Wang, Angew. Chem. Int. Ed. 60 (2021) 22547-22553. DOI:10.1002/anie.202109767 |
| [31] |
X. Tang, M. Hong, L. Falivene, et al., J. Am. Chem. Soc. 138 (2016) 14326-14337. DOI:10.1021/jacs.6b07974 |
| [32] |
H.Z. Fan, X. Yang, J.H. Chen, et al., Angew. Chem. Int. Ed. 61 (2022) e202117639. DOI:10.1002/anie.202117639 |
| [33] |
W. Xiong, W. Chang, D. Shi, et al., Chemistry 6 (2020) 1831-1843. DOI:10.1016/j.chempr.2020.06.003 |
| [34] |
Y. Yu, L.M. Fang, Y. Liu, X.B. Lu, ACS Catal. 11 (2021) 8349-8357. DOI:10.1021/acscatal.1c01376 |
| [35] |
J. Yuan, W. Xiong, X. Zhou, et al., J. Am. Chem. Soc. 141 (2019) 4928-4935. DOI:10.1021/jacs.9b00031 |
| [36] |
Z. Wang, R. Yang, G. Xu, T. Liu, Q. Wang, ACS Sustain. Chem. Eng. 10 (2022) 4529-4537. DOI:10.1021/acssuschemeng.1c08408 |
| [37] |
J.B. Zhu, E.Y.X. Chen, Angew. Chem. Int. Ed. 58 (2019) 1178-1182. DOI:10.1002/anie.201813006 |
| [38] |
E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven, W. Eevers, Polym. Degrad. Stabil. 179 (2020) 109241. DOI:10.1016/j.polymdegradstab.2020.109241 |
| [39] |
X. Zhang, M. Fevre, G.O. Jones, R.M. Waymouth, Chem. Rev. 118 (2018) 839-885. DOI:10.1021/acs.chemrev.7b00329 |
| [40] |
A.B. Kremer, P. Mehrkhodavandi, Coord. Chem. Rev. 380 (2019) 35-57. DOI:10.1016/j.ccr.2018.09.008 |
| [41] |
L. Shen, Z. Chen, Q. Zheng, et al., ACS Catal. 11 (2021) 12833-12839. DOI:10.1021/acscatal.1c04354 |
| [42] |
J. Wu, L. Shen, S. Duan, et al., Angew. Chem. Int. Ed. 59 (2020) 13871-13878. DOI:10.1002/anie.202004174 |
| [43] |
J. Wu, L. Shen, Z.N. Chen, et al., Angew. Chem. Int. Ed. 59 (2020) 10421-10425. DOI:10.1002/anie.202002403 |
| [44] |
Y. Shen, Q. Zheng, H. Zhu, T. Tu, Adv. Mater. 32 (2020) 1905950. DOI:10.1002/adma.201905950 |
| [45] |
G. Kale, R. Auras, S.P. Singh, J. Polym. Environ. 14 (2006) 317-334. DOI:10.1007/s10924-006-0015-6 |
| [46] |
P. McKeown, M.D. Jones, Sustain. Chem. 1 (2020) 1-22. DOI:10.3390/suschem1010001 |
| [47] |
T. Tsukegi, T. Motoyama, Y. Shirai, H. Nishida, T. Endo, Polym. Degrad. Stabil. 92 (2007) 552-559. DOI:10.1016/j.polymdegradstab.2007.01.009 |
| [48] |
H. Nishida, T. Mori, S. Hoshihara, et al., Polym. Degrad. Stabil. 81 (2003) 515-523. DOI:10.1016/S0141-3910(03)00152-6 |
| [49] |
L. Dai, R. Liu, C. Si, Green Chem. 20 (2018) 1777-1783. DOI:10.1039/C7GC03863A |
| [50] |
C.S.M. Pereira, V.M.T.M. Silva, A.E. Rodrigues, Green Chem. 13 (2011) 2658-2971. DOI:10.1039/c1gc15523g |
| [51] |
M. Hofmann, C. Alberti, F. Scheliga, R.R.R. Meißner, S. Enthaler, Polym. Chem. 11 (2020) 2625-2629. DOI:10.1039/D0PY00292E |
| [52] |
F.A. Leibfarth, N. Moreno, A.P. Hawker, J.D. Shand, J. Polym. Sci. Part A 50 (2012) 4814-4822. DOI:10.1002/pola.26303 |
| [53] |
M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu, ACS Sustain. Chem. Eng. 6 (2018) 15127-15134. DOI:10.1021/acssuschemeng.8b03591 |
| [54] |
P. McKeown, M. Kamran, M.G. Davidson, et al., Green Chem. 22 (2020) 3721-3726. DOI:10.1039/D0GC01252A |
| [55] |
P. McKeown, L.A. Roman-Ramirez, S. Bates, J. Wood, M.D. Jones, ChemSusChem 12 (2019) 5233-5238. DOI:10.1002/cssc.201902755 |
| [56] |
J. Payne, P. McKeown, O. Driscoll, et al., Polym. Chem. 12 (2021) 1086-1096. DOI:10.1039/D0PY01519A |
| [57] |
R. Petrus, D. Bykowski, P. Sobota, ACS Catal 6 (2016) 5222-5235. DOI:10.1021/acscatal.6b01009 |
| [58] |
R. Yang, G. Xu, C. Lv, et al., ACS Sustain. Chem. Eng. 8 (2020) 18347-18353. DOI:10.1021/acssuschemeng.0c07595 |
| [59] |
E.L. Whitelaw, M.G. Davidson, M.D. Jones, Chem. Commun. 47 (2011) 10004-10006. DOI:10.1039/c1cc13910j |
| [60] |
J.M. Payne, G. Kociok-Köhn, E.A.C. Emanuelsson, M.D. Jones, Macromolecules 54 (2021) 8453-8469. DOI:10.1021/acs.macromol.1c01207 |
| [61] |
R. Petrus, P. Sobota, Organometallics 31 (2012) 4755-4762. DOI:10.1021/om300321h |
| [62] |
Y. Wang, W. Zhao, X. Liu, D. Cui, E.Y.X. Chen, Macromolecules 45 (2012) 6957-6965. DOI:10.1021/ma3007625 |
| [63] |
B. Orhan, M.J.L. Tschan, A.L. Wirotius, et al., ACS Macro Lett. 7 (2018) 1413-1419. DOI:10.1021/acsmacrolett.8b00852 |
| [64] |
Z. Dai, Y. Sun, J. Xiong, X. Pan, J. Wu, ACS Macro Lett. 4 (2015) 556-560. DOI:10.1021/acsmacrolett.5b00209 |
| [65] |
C. Shi, L.T. Reilly, V.S. Phani Kumar, et al., Chemistry 7 (2021) 2896-2912. DOI:10.1016/j.chempr.2021.10.004 |
| [66] |
A. Duda, S. Penczek, Macromolecules 23 (1990) 1636-1639. DOI:10.1021/ma00208a012 |
| [67] |
M. Hong, E.Y.X. Chen, Green Chem. 19 (2017) 3692-3706. DOI:10.1039/C7GC01496A |
| [68] |
Y. Zhang, Y. Qi, Y. Yin, et al., ACS Sustain. Chem. Eng. 8 (2020) 2865-2873. DOI:10.1021/acssuschemeng.9b06987 |
| [69] |
R. Yang, G. Xu, B. Dong, H. Hou, Q. Wang, Macromolecules 55 (2022) 1726-1735. DOI:10.1021/acs.macromol.1c02085 |