 2023, Vol. 34
2023, Vol. 34 


Covalent inhibitors have received increased attention in recent years as an important complementary or alternative drugs to their non-covalent counterparts due to their long-acting nature and cumulative enhanced potency on the same target(s) [1]. Natural products are a quintessential resource for developing covalent drugs, even now as the research paradigm is shifting from an accidental discovery to a rational design phase [2]. Innovative ideas are often inspired by innate covalent inhibitors, formed when an endogenous electrophile reacts with protein targets [3,4]. As the potential targets of nucleophilic residues, activated cysteine and serine residues are expected to be the most attacked binding-sites by Michael acceptors, aziridines, epoxides, and β-lactams [5]. Targeting nucleophilic lysine residues can also present a viable approach to irreversible conjugation [6–8]. A marine active natural product, manoalide, which is a sesterterpenoid antibiotic, has been proposed to inhibit phospholipase A2 (PLA2) by initial imine formation by binding to two lysine residues at Lys6 and Lys79 via two masked aldehyde lactols [9–14]. Wortmannin, an antibiotic produced by Penicillium wortmanni, has been shown to irreversibly bind to catalytic Lys802 or Lys833 in the active-site of phosphoinositide 3-kinases (PI3K), depending on the furan ring (C20), followed by ring opening to give the enamine species [15–18]. Early covalent inhibitors all depended on natural products. However, the design of modern targeted covalent inhibitors has also required a degree of serendipity, leading to the eventual discovery of novel covalent binding mechanisms.
Phenylpropionyl iridoids are a notable natural product with a cyclopentane ring and hemiacetal structural features. Based on the unique structural features and complex stereo configurations, these iridoids exhibit multipole desirable biological activities [19]. Genipin (GNP) is a geniposide-derived aglycone found primarily in the fruits of Gardenia and American Gardenia [20]. It has long been used in traditional oriental medicine to prevent and treat cancer, inflammation, diabetes, and neurological disorders [21,22]. Previous studies have shown that oral geniposide can be hydrolysed by most intestinal bacteria, and GNP is formed after deglycosylation [23]. Moreover, GNP is generally described as a strong cross-linker with less cytotoxicity in bioactive materials than glutaraldehyde [24]. The cross-linking mechanism is thought to occur between GNP and primary amine-containing molecules. Interestingly, GNP has been reported to inhibit uncoupling protein 2 (UCP2) in mitochondria via a non-crosslinking mechanism and thus block UCP2-mediated proton leak [25]. C14-OH in GNP is suggested as the key functional group responsible for its bioactivity, including inducing cell apoptosis by inhibiting UCP2 and reducing reactive oxygen species (ROS) [26]. However, GNP remains unpopular in clinical use since the detailed mechanisms of action are poorly understood.
UCP2 is located in the inner mitochondrial membrane and is highly homologous to other UCP subfamily members such as UCP3 and UCP1 [27]. The best understood non-shivering thermogenic system in brown adipose tissue which has evolved to protect the body from hypothermia, involves UCP1, a key regulator of cold-mediated thermogenesis. In contrast, UCP2 is expressed in many organs and tissues in the body. UCP2 participates in the body's thermogenesis through the electrochemical gradient of dissipating protons, and regulates not only mitochondrial ATP production, but also the generation of reactive oxygen species (ROS). Evidence highlighting the role of UCP2 in a broad range of physiological and pathological processes continues to accumulate [28]. The coupling/uncoupling state is related to the permeability of the inner mitochondrial membrane [29,30]. UCP2 transfers protons directly from the intermembranous space into the matrix and fatty acid anions in the opposite direction, reducing the proton gradient and decoupling oxidative phosphorylation, thereby controlling the ATP/ADP ratio and the inner mitochondrial membrane proton gradient [31]. The evidence indicates that the expression of UCP2 in the liver has the same trend as the body temperature of mice [32]. Surprisingly, mitochondrial thermogenesis can reach temperatures close to 50 ℃ [33]. Due to the fact that heat can be rapidly dissipated into the extracellular environment, the mitochondria thermometer or molecule probe of UCP should be tightly attached to the organelle to detect the changes in heat [34].
Although GNP acts as a specific inhibitor of UCP2-mediated proton transport [35,36], little research has been conducted to determine the precise antipyretic mechanisms involving GNP and UCP2. In this paper, two alkynyl-modified GNP (Alk-GNP) probes with different modifications were designed and synthesised. A series of chemical and biological methods were used to identify the mechanism underlying the antipyretic action of GNP by intraperitoneal injection of Alk-GNP probes in mice. Subsequently, a novel irreversible reaction model for targeting lysine in UCP2 proton leak was proposed. The hemiacetal structure in the phenylpropionyl iridoid of GNP undergoes a ring-opening and condensation process, and thus undergoes temperature-sensitive binding to the ɛ-amine of lysine in UCP2 under physiological conditions, reducing proton transport function and thermogenesis.
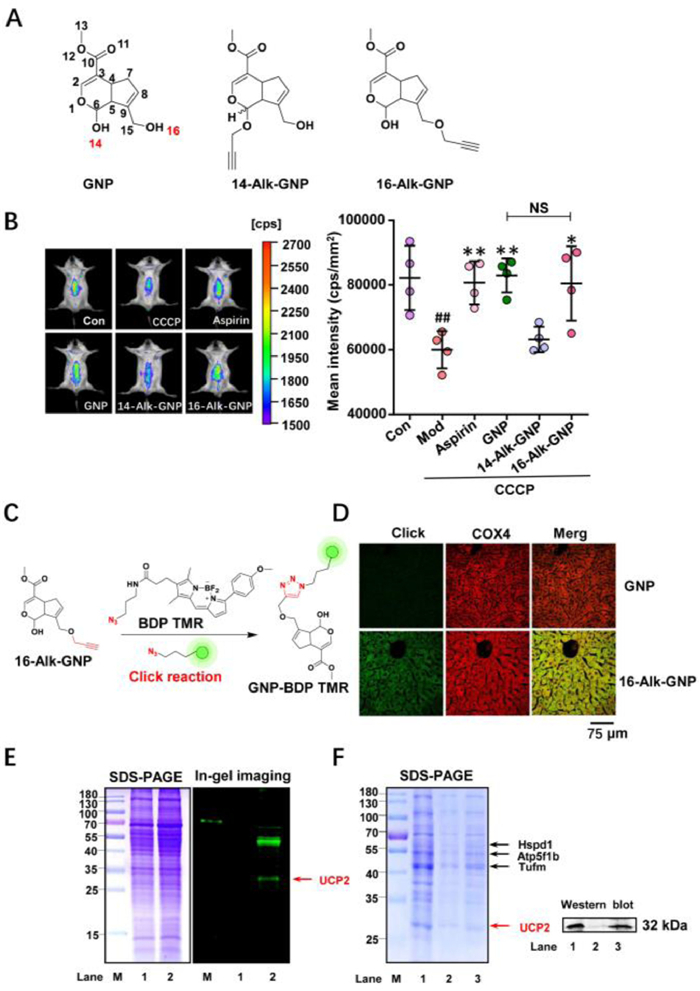
GNP targets mitochondrial UCP2 and exerts an antipyretic effect. To reveal the antipyretic mechanism of GNP, two alkynylation probes, 14-Alk-GNP and 16-Alk-GNP, were prepared at different hydroxyl sites (Fig. 1A and Figs. S1–S8 in Supporting information), and the antipyretic effects on mouse liver were subsequently evaluated via a thermosensitive rhodamine B (RhB)-derived fluorogenic probe (RhBIV) [37]. A high temperature model in mice was induced using carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (7 mg/kg, i.p.), a mitochondrial electron transport chain inhibitor, and aspirin (300 mg/kg, p.o.) as a positive control. The animal experiments were approved by the Animal Ethics Committee, Guangxi University of Chinese Medicine, China (No. DW20200711-098). The result showed that the 16-Alk-GNP probe (20 mg/kg, i.p.), which retained the hemiacetal structure, had the same antipyretic effect as GNP (20 mg/kg, i.p.); the dose dependent nature of the antipyretic effect is shown in Fig. S9 (Supporting information). In contrast, 14-Alk-GNP (20 mg/kg, i.p.) lost its original activity (Fig. 1B), indicating that the hemiacetal structure and C14-OH might be responsible for the activity. To further investigate the distribution of GNP in liver tissues, liver sections were incubated with BDP TMR azide following intraperitoneal injection of 16-Alk-GNP or GNP for target imaging (Fig. 1C). The imaging of the 16-Alk-GNP probe (pseudo-green) showed that it appeared to partially co-localize with the mitochondrial protein COX4; COX4 is bound by primary antibody coupled with AlexaFluor®594-conjugated secondary antibody (pseudo-red), thus colocalisation is detected as a merged yellow. The Pearson coefficient (PC) was approximately 0.924 (Fig. 1D).

|
Download:
|
| Fig. 1. UCP2 is identified as an antipyretic target on mitochondria for GNP. (A) The chemical structures of GNP, 14-AlK GNP, and 16-AlK-GNP probe. (B) The antipyretic effect of GNP and its probes, detected via a thermosensitive RhBIV probe on CCCP (7 mg/kg) induced fever model (left panel). The statistical analysis of the fluorescence intensity is represented as the mean ± SD (right panel). Comparisons between two groups were carried out by Student's t-test (NS, no significant), and one-way analysis of variance (ANOVA) was used to identify differences among groups. All groups except the Con group were treated with CCCP for the establishment of an induced high temperature model. ##P < 0.01 vs. Con, **P < 0.01, *P < 0.05 vs. CCCP (n = 4). (C) The chemical reaction process of 16-AlK-GNP with N3-BDP TMR. (D) The co-localization assay between the mitochondrial COX4 protein and 16-AlK-GNP probe on liver tissue sections. (E) The SDS-PAGE and in-gel imaging assay of mice liver mitochondria lysate after intraperitoneal injection of 20 mg/kg GNP (Lane 1) or 20 mg/kg 16-AlK-GNP (Lane 2). (F) SDS-PAGE and western blot assay for target fishing on mice liver mitochondria. Lane M, Marker; Lane 1 shows the mitochondria lysate; Lane 2 represents the negative control with blank beads; Lane 3 shows the captured protein by the 16-AlK-GNP probe. | |
{kind=link}
Subsequently, the potential targets of GNP on mitochondria were investigated by in-gel imaging assay. As shown in Fig. 1E, several distinct bands of 16-Alk-GNP labelled proteins were detected in the mouse liver microsphere fraction. The targets were then captured using N3-tagged magnetic microspheres according to previously reported methods (Fig. 1F, left panel) [38]. When compared to the control beads (lane 2), several potential proteins were found to be enriched by 16-Alk-GNP linked beads (lane 3). The captured proteins were then released and identified by Triple-quad Ion-trap and Orbitrap fusion (Thermo Scientific, USA) and KEGG functional annotation was performed. Among these proteins, UCP2 (approximately 32 kDa) was the most likely target. The identification was also corroborated by the western blot analysis (Fig. 1F, right panel). Unlike mitochondrial ATP synthase (ATP5f1b), UCP2 can separate oxidative phosphorylation from ATP synthesis and the return transfer of protons from the outer to the inner mitochondrial membrane dissipates energy as heat [39]. Next, the effects of GNP on mitochondrial membrane potential and thermogenesis were evaluated at the cellular level.
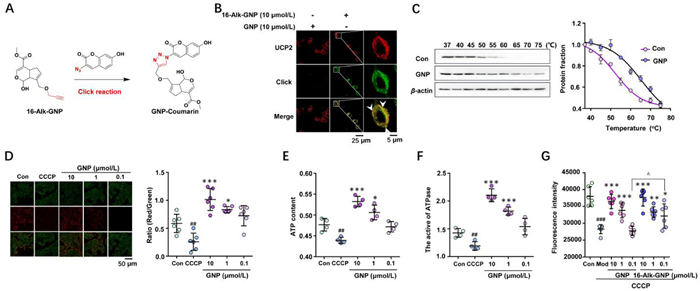
GNP blocks the return transfer of protons into the inner mitochondrial membrane and reduces thermogenesis. A click reaction between 16-Alk-GNP and the N3-coumarin tag was designed to generate a fluorescent derivative for tracking GNP at the cellular level (Fig. 2A). The 16-Alk-GNP probe (pseudo-green) and UCP2 protein stained by primary antibody coupled with AlexaFluor®594-secondary antibody (pseudo-red) colocalised well in the cytoplasm of HepG2 cells (PC = 0.831), while almost no fluorescence was observed in the unmodified GNP group (PC = 0.281) (Fig. 2B). In addition, the interaction between GNP and UCP2 was evaluated using a cellular thermal shift assay (CETSA) with a gradient temperature treatment (37–75 ℃) for 3 min, followed by a western blot analysis. As shown in Fig. 2C, GNP treatment improved the thermal stability of UCP2 in cell lysates in a dose-dependent manner compared to the control group.

|
Download:
|
| Fig. 2. GNP targets mitochondrial UCP2 to affect its function. (A) The click reaction process of 16-AlK GNP with an N3-coumarin tag. (B) Co-localization of 16-Alk-GNP probe (pseudo-green) with UCP2 (pseudo-red) in HepG-2 cells. (C) The cellular thermal shift assay (CETSA) of UCP2 treated with GNP (10 µmol/L) in HepG-2 cell lysates detected by western blot. The statistics assay was performed by the UCP2 staining of the control or GNP group, with normalization against the levels of β-actin (n = 3). (D) The effects of GNP on mitochondrial membrane potential (MMP), The Red/Green fluorescence images of MMP detected by the JC-1 probe (n = 6). ATP content (E) and the activity of mitochondrial complex Ⅴ (F) in HepG2 cells (n = 4). (G) The effects of GNP and 16-Alk-GNP on the mitochondrial temperature in HepG2 cells monitored via RhBIV probe. The values of the image were expressed as mean ± SD. One-way analysis of variance (ANOVA) was used to identify differences among groups. The comparison between the two groups was carried out by Student's t-test. ##P < 0.01, ###P < 0.001 vs. Con; ***P < 0.001, **P < 0.01, *P < 0.05 vs. CCCP; △P < 0.05 vs. GNP (n = 6). | |
{kind=link}
To investigate GNP's effect on UCP2, a commercial JC-1 probe was used to detect mitochondrial membrane potential (MMP) changes induced by CCCP (50 µmol/L), with or without GNP intervention. As shown in Fig. 2D, GNP (red/green) increased MMP in HepG-2 cells in a dose-dependent manner (0.1–10 µmol/L). The proton kinetic energy drove protons back to the substrate through complex Ⅴ (ATP synthase), which promoted the phosphorylation of ADP to ATP in the presence of phosphorus. As shown in Figs. 2E and F, GNP increased the ATP content and improved the activity of ATP synthase. The changes in mitochondrial temperature were then assessed using RhBIV probe (Fig. 2G). It was found that both GNP and Alk-GNP probes reversed CCCP-induced mitochondrial temperature increase, with GNP modified at the C16-OH group showing a greater inhibitory effect at the low concentration (0.1 µmol/L), alkylation may have prevented GNP from self-polymerising via C16-OH groups. Based on these results, we hypothesised that GNP may block the return transfer of protons into the inner mitochondrial membrane and attenuate the thermogenesis process by covalently binding with UPC2 in HepG2 cells.
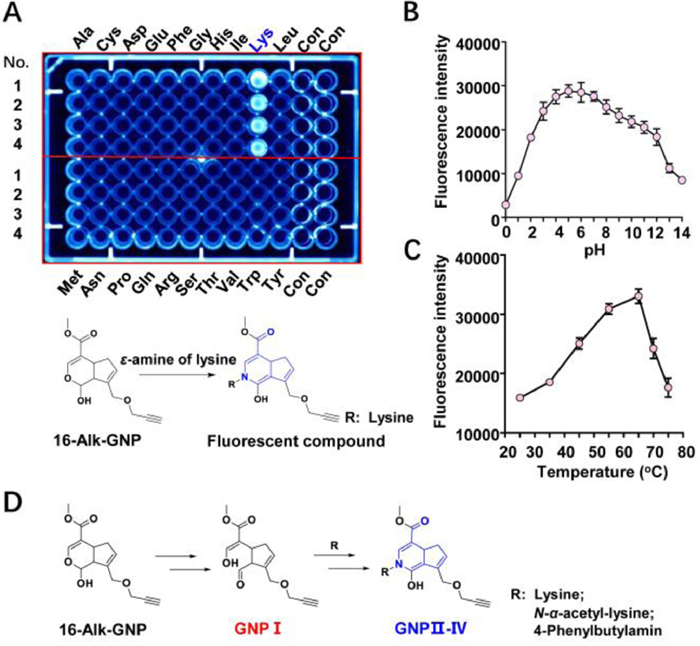
The hemiacetal structure of GNP specifically attacks the ɛ-amine on lysine. Normally, only low-abundance cysteine residues are proximal to druggable binding sites. Other nucleophilic residues chosen for nucleophilic addition include lysine, serine, and tyrosine [40]. To investigate the irreversible binding mode of GNP to amino acid residues on UCP2, 20 free amino acids were selected to react with GNP or 16-Alk-GNP probe in physiological conditions. As shown in Fig. 3A (upper panel), lysine can significantly react with the 16-Alk-GNP probe to form a fluorescence product (Fig. S10 in Supporting information), while unmodified GNP can also form a cross-linked blue pigment with lysine (Figs. S11 and S12 in Supporting information). Since 16-Alk-GNP lacks the probe missed the C-16 hydroxyl group, the reaction was assumed to be a condensation reaction involving the C-14 hemiacetal structure in GNP and the ɛ-amine of lysine (Fig. 3A, down panel). Such a reaction has previously been reported to produce a dietary gardenia blue pigment derived from GNP and tyrosine [41]. Geniposide was hydrolysed to GNP by microbial glycosidases, and GNP was spontaneously converted to a dialdehyde intermediate. The dialdehyde then covalently bound to the primary amine residues of free amino acids [42]. The polymer fragments were used to infer the binding mode of GNP to lysine and other amino acids [43]; howerer, the detailed data is scarce.

|
Download:
|
| Fig. 3. The hemiacetal group of GNP selectivity reacts with the ɛ-amine of lysine. (A) Cross-linking reactions of 16-Alk-GNP (1 mmol/L) with 20 free amino acids (1 mmol/L), incubated in physiological conditions for 45 min. The investigation of optimum reaction conditions of pH (B) and temperature (C) for the reaction between 16-Alk-GNP (1 mmol/L) and lysine (1 mmol/L). (D) Proposed mechanism of the hemiacetal condensation between 16-Alk-GNP and ɛ-amine compounds in physiological conditions. | |
{kind=link}
To avoid the cross-linking effect and gain a better insight into the novel mechanism, the 16‑hydroxyl group in GNP was first sealed by alkynylation. The lysine residues contain a potentially nucleophilic primary amine that can covalently bond with an electrophilic functional "warhead, " but this irreversible bonding is subject to specific constraints [44]. Therefore, the optimum temperature and pH for the reaction between 16-Alk-GNP and lysine were investigated. The results showed that condensation could work well under physiological pH conditions (Fig. 3B). Interestingly, the reaction efficiency gradually as the temperature was increased from 25 ℃ to 65 ℃ but decreased as the temperature was raised further from 65 ℃ to 75 ℃ (Fig. 3C).
Generally, selective irreversible inhibitors must operate through two steps. Firstly, they bind reversibly to non-covalent complexes and subsequently undergo covalent bond formation, which is only fast once the reversible complex has formed [45]. It was therefore important to know how to trigger the ring-opening condensation reaction of hemiacetal in physiological conditions. Aldehydes have commonly been reported to modify lysine residues in proteomics [46]. Due to its unstable hemiacetal structure, 1-Alk-GNP was synthesised to capture the enol structure of GNP Ⅰ (Figs. S13–S17 in Supporting information). The condensation process from GNP Ⅰ with ɛ-amine compounds (lysine, N-α-acetyl-lysine or 4-phenylbutylamine) to the GNP Ⅱ–Ⅳ end products was then monitored. The chemical interaction mode between them is suggested in Fig. 3D and Fig. S18 (Supporting information), to involve an electrophilic aldehyde group, which is then cross-linked with the ɛ-amine group. The detailed information is shown in supporting data (Figs. S19–S28 in Supporting information).
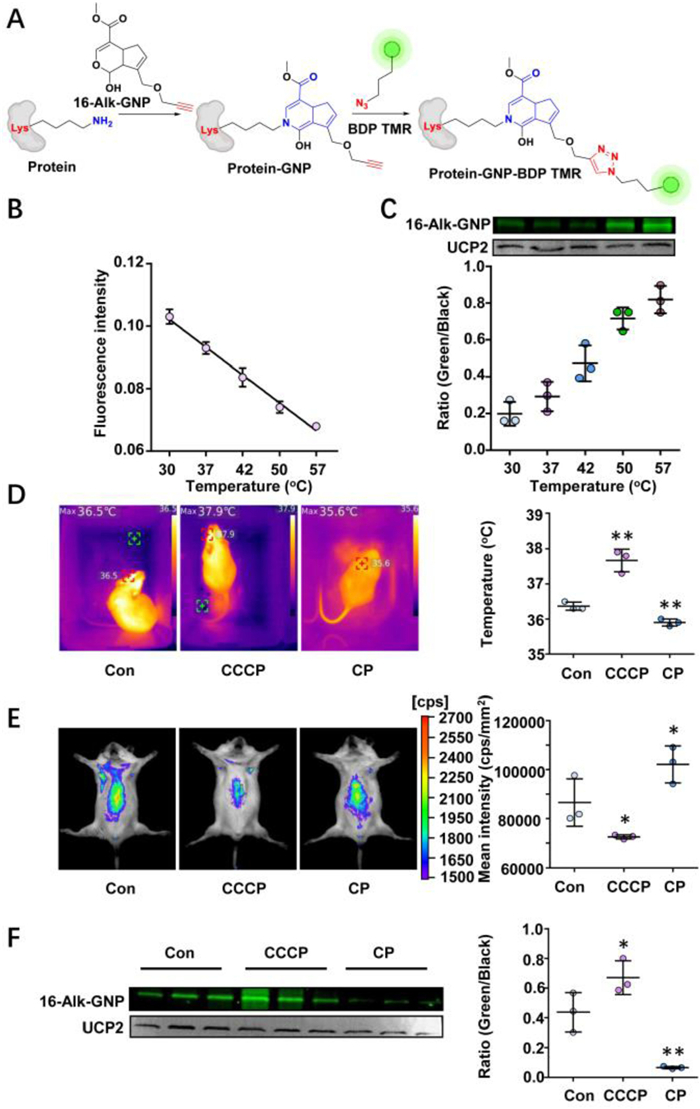
Conjugation of the hemiacetal group and ɛ-amine on lysine showed a temperature responsiveness. The conjugation efficiency between GNP and UCP was investigated both in vitro and in vivo. The bioorthogonal reaction strategy based on protein lysine ε-amine group and 16-Alk-GNP hemiacetal is shown in Fig. 4A. Liver mitochondria were artificially incubated for 1 h with 16-Alk-GNP (10 µmol/L) at the specified temperatures (30–57 ℃), and the temperature responsiveness was monitored simultaneously with 1 µmol/L RhBIV probe (Fig. 4B). An in-gel imaging assay was used to determine the conjugation efficiency of 16-Alk-GNP with UCP2. As shown in Fig. 4C, the fluorescence intensity was gradually increased with temperature response.

|
Download:
|
| Fig. 4. The irreversible conjugation of GNP and UCP2 demonstrates temperature sensitivity both in vitro and in vivo. (A) A bioorthogonal reaction strategy based on protein lysine ɛ-amine group. (B) The changes in temperature response on the extracted liver mitochondria detected by a thermosensitive RhBIV probe (n = 5). (C) The cross-linking efficiency of 16-Alk-GNP to UCP2 protein with temperature response, shown by an in-gel imaging assay and normalised via western blot (n = 3). (D) The fever model established by CCCP and the low-temperature model-performed by CP. The body temperature was monitored with an infrared temperature imager. (E) The mice's liver temperature for fever and low-temperature models monitored by a RhBIV probe. (F) In-gel imaging assay used to trace the conjugation efficiency between the 16-Alk-GNP probe and UCP2 protein in mice liver, and western blot performed to normalize the UCP2 content. The values were the mean ± SD. P < 0.05, **P < 0.01 and #P < 0.05, ##P < 0.01 were respectively indicated for raising and lowering the temperature vs. Con group (n = 3). | |
{kind=link}
To explore whether the characteristics actually exist in vivo, CCCP (7 mg/kg, i.p.) was administered to induce fever in mice, while chlorpromazine (CP) (8 mg/kg, i.p.) was used to induce low temperature. Afterwards, 16-Alk-GNP (20 mg/kg, i.p.) was administered intraperitoneally for 30 min. The changes in body temperature were investigated via an infrared temperature imager (UTi260K, China) in mice. As shown in Fig. 4D and Fig. S29 (Supporting information), CCCP significantly increased the body temperature, whereas the CP group obtained the expected temperature decrease. Meanwhile, monitoring liver temperature yielded the same results confirmed by RhBIV imaging (Fig. 4E). Noteworthy, the in-gel imaging results for liver mitochondria showed that the CCCP treatment facilitated the conjugation efficiency of the 16-Alk-GNP to UCP2 (P < 0.05), while the intervention of CP significantly prevented the binding process (P < 0.01) (Fig. 4F).
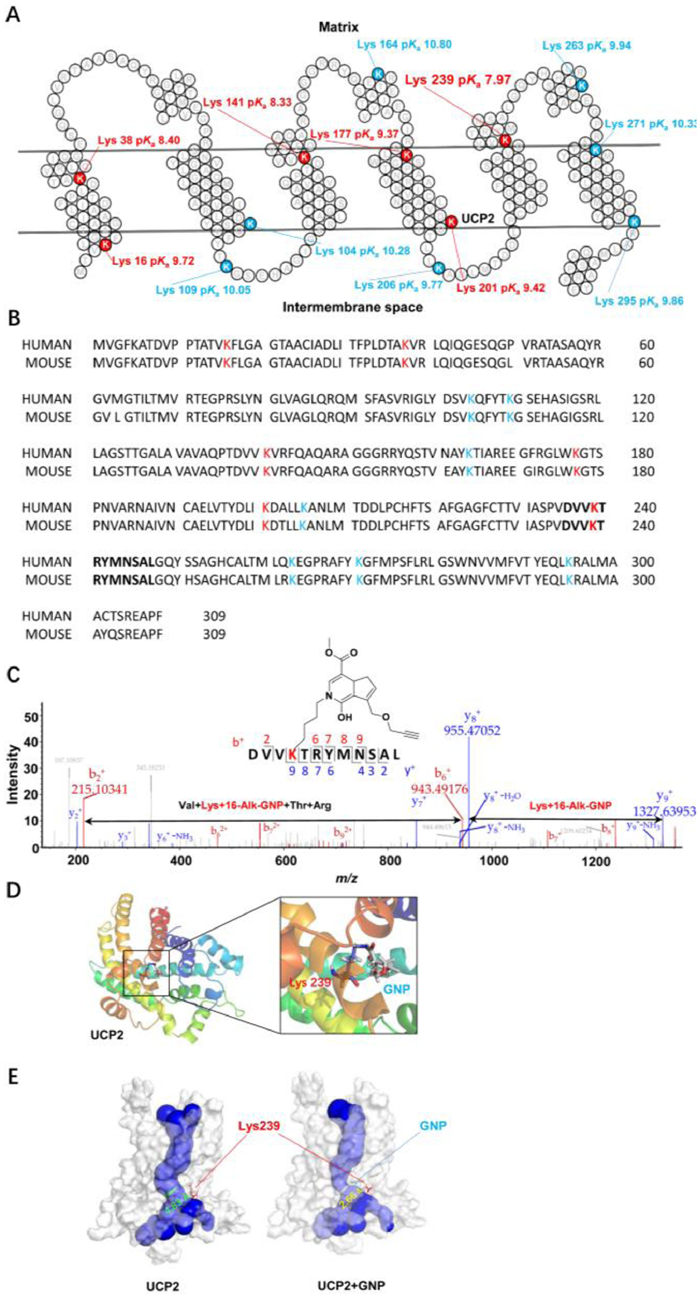
The irreversible binding of GNP to lysine in the proton leak cavity on UCP2 induces the antipyretic effect. The surface ɛ-amino group of lysine residues on target proteins is the most difficult for the nucleophilic reaction because they are largely exposed to physiological pH (7.4) and are unavailable to nucleophiles [47]. To successfully target lysine residues, different ionization states of various amino acids must be used to decrease pKa values [48]. Lower orders of pKa in the active free lysine residues promote irreversible binding. Hence, the pKa values of lysine in UCP2 were predicted using Propka 3.1 software and the results are shown in Fig. 5A. It was previously shown that GNP inhibits UCP1 at low concentrations (50 µmol/L) by binding to lysine (Lys198) and arginine, and modifies cysteine (Cys188) at high concentrations (1 mmol/L) [29]. The homology between human UCP1 and UCP2 is only 56.9%. However, the sequence identity of UCP2 between humans and mice is higher than 96% and all lysines are highly conserved, which suggests they have the same covalent binding mode (Fig. 5B). Compared with most surface lysines on UCP2 (blue), the lysines in the proton leak pipeline (red) harbor a more suitable pKa for nucleophilic attack.

|
Download:
|
| Fig. 5. The distribution and pKa prediction of lysine on UCP2 and the covalent binding mode with GNP. (A) Schematic diagram of the UCP2 protein (PDB ID: 2LCK) with altered pKa of internal lysine compared to exposed lysine residues. The pKa values of the key lysine residues are highlighted in red or blue (Propka 3.1). (B) Sequence homology alignment of UCP2 between humans and mice. (C) LC-MS/MS analysis of the recombinant UCP2 protein incubated with 16-Alk-GNP for 12 h. 16-Alk-GNP binds to the Lys239 residue of the UCP2 fragment. (D) Covalent docking model between hemiacetal in GNP and the ɛ-amino group of Lys239. Cartoon representation and mesh protein surface were generated using PyMOL Molecular Graphics System, Version 3.2 Schrçdinger. (E) 3D structure simulation of UCP2 protein on proton leak cavity for irreversible binding with or without GNP, built using Caver 3.03 plugin. | |
{kind=link}
To further reveal the binding mode of GNP with UCP2, purified mouse recombinant UCP2 was incubated with16-Alk-GNP and then identified by protein MS/MS profiles. As shown in Fig. 5C, for the b- and y-fragment ions of the UCP2 peptide, the m/z shift was 372.1690 between the y8+ and y9+ fragment ions, consistent with the mass of Lys239-labelled 16-Alk-GNP (372.1691). In addition, the m/z shift 728.3884 between the b2+ and b6+ fragment ions agreed with the mass of Val238-Lys239-labelled 16-Alk-GNP-Thr240-Arg241 (728.3868). Hence, covalent binding was identified at Lys239. Moreover, this covalent binding may also occur at other lysine sites, such as Lys164 (Fig. S30 in Supporting information).
Subsequently, the Lys239 on UCP2 (pKa 7.97) was selected as an example for covalent docking with GNP in the binding mode, and a covalent docking simulation between hemiacetal in GNP and ɛ-amine of Lys239 on UCP2 was suggested (Fig. 5D). The result demonstrated that irreversible binding blocks the proton leak pipeline in UCP2 from 4.83 to 2.66 Å (Caver3.03 plugin) (Fig. 5E). This covalent binding narrows the proton leak tunnel, leading to a decrease in the reduction of H+ leakage via UCP2. By preventing proton transfer from the intermembranous space into the matrix, the conjugation of GNP to UCP2 increases the MMP, and changes the reflux of protons via ATP synthase, thus improving the efficiency of ATP production. By blocking the decoupling oxidative phosphorylation, the binding ultimately exerts an antipyretic effect.
In summary, the irreversible binding of GNP's hemiacetal group to Lys239 in UCP2 plays a key role in the proton leak cavity. The nucleophilic attack with temperature response reduces mitochondrial thermogenesis by blocking the efficiency of proton dissipation. Besides demonstrating the antipyretic mechanism, this study provides an expanded understanding of the interaction between hemiacetal groups and ɛ-amine. It is anticipated to be used as a specific modification tool for the ɛ-amine of lysine on proteins, which can be applied in bioorthogonal reactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (No. 81973449) and the National Key Research and Development Program of China (Nos. 2018YFC1704800 and 2018YFC1704805).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108203.
| [1] |
H. Kim, Y.S. Hwang, M. Kim, S.B. Park, RSC Med. Chem. 12 (2021) 1037-1045. DOI:10.1039/d1md00068c |
| [2] |
F. Sutanto, M. Konstantinidou, A. Domling, RSC Med. Chem. 11 (2020) 876-884. DOI:10.1039/d0md00154f |
| [3] |
W. Lei, F.K. Shen, N.W. Chang, et al., Chin. Chem. Lett. 32 (2021) 190-193. DOI:10.1016/j.cclet.2020.10.049 |
| [4] |
H.K. Xu, X. Qin, Y.P. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2001-2004. DOI:10.1016/j.cclet.2021.09.071 |
| [5] |
S. De Cesco, J. Kurian, C. Dufresne, et al., Eur. J. Med. Chem. 138 (2017) 96-114. DOI:10.1016/j.ejmech.2017.06.019 |
| [6] |
J. Pettinger, K. Jones, M.D. Cheeseman, Angew. Chem. Int. Ed. 56 (2017) 15200-15209. DOI:10.1002/anie.201707630 |
| [7] |
R.M. Reja, W. Wang, Y. Lyu, et al., J. Am. Chem. Soc. 144 (2022) 1152-1157. DOI:10.1021/jacs.1c12702 |
| [8] |
D. Quach, G. Tang, J. Anantharajan, et al., Angew. Chem. Int. Ed. 60 (2021) 17131-17137. DOI:10.1002/anie.202105383 |
| [9] |
R.A. Deems, D. Lombardo, B.P. Morgan, et al., Biochim. Biophys. Acta 917 (1987) 258-268. DOI:10.1016/0005-2760(87)90130-5 |
| [10] |
L.J. Reynolds, E.D. Mihelich, E.A. Dennis, J. Biol. Chem. 266 (1991) 16512-16517. DOI:10.1016/S0021-9258(18)55330-8 |
| [11] |
D. Lombardo, E.A. Dennis, J. Biol. Chem. 260 (1985) 7234-7240. DOI:10.1016/S0021-9258(17)39598-4 |
| [12] |
K.B. Glaser, R.S. Jacobs, Biochem. Pharmacol. 36 (1987) 2079-2086. DOI:10.1016/0006-2952(87)90134-1 |
| [13] |
K.B. Glaser, M.S. de Carvalho, R.S. Jacobs, et al., Mol. Pharmacol. 36 (1989) 782-788. |
| [14] |
I.D. Bianco, M.J. Kelley, R.M. Crowl, E.A. Dennis, Biochim. Biophys. Acta 1250 (1995) 197-203. DOI:10.1016/0167-4838(95)00051-U |
| [15] |
T. Frew, G. Powis, M. Berggren, et al., Anticancer Drug Des. 10 (1995) 347-359. DOI:10.1016/0378-8741(95)01246-A |
| [16] |
T. Frew, G. Powis, M. Berggren, et al., Anticancer Res. 14 (1994) 2425-2428. DOI:10.1007/BF02303619 |
| [17] |
M.P. Wymann, G. Bulgarelli-Leva, M.J. Zvelebil, et al., Mol. Cell. Biol. 16 (1996) 1722-1733. DOI:10.1128/MCB.16.4.1722 |
| [18] |
G. Powis, R. Bonjouklian, M.M. Berggren, et al., Cancer Res. 54 (1994) 2419-2423. DOI:10.1158/1538-7445.am2016-ct157 |
| [19] |
J. Cao, H. Yu, Y. Wu, X. Wang, Mini Rev. Med. Chem. 19 (2019) 292-309. DOI:10.2174/1389557518666181026091449 |
| [20] |
C.H. Chang, J.B. Wu, J.S. Yang, et al., J. Food Sci. 82 (2017) 3021-3028. DOI:10.1111/1750-3841.13955 |
| [21] |
M.K. Shanmugam, H. Shen, F.R. Tang, et al., Pharmacol. Res. 133 (2018) 195-200. DOI:10.1016/j.phrs.2018.05.007 |
| [22] |
C.C. Li, C.Y. Hsiang, H.Y. Lo, et al., Food Chem. Toxicol. 50 (2012) 2978-2986. DOI:10.1016/j.fct.2012.05.054 |
| [23] |
T. Akao, K. Kobashi, M. Aburada, Biol. Pharm. Bull. 17 (1994) 1573-1576. DOI:10.1248/bpb.17.1573 |
| [24] |
Y. Liu, Z. Cai, L. Sheng, et al., Carbohydr. Polym. 215 (2019) 348-357. DOI:10.3390/mi10050348 |
| [25] |
C.Y. Zhang, L.E. Parton, C.P. Ye, et al., Cell Metab. 3 (2006) 417-427. DOI:10.1016/j.cmet.2006.04.010 |
| [26] |
Y. Yang, Y. Yang, J. Hou, et al., PLoS One 11 (2016) e0147026. DOI:10.1371/journal.pone.0147026 |
| [27] |
A. Matthias, K.B. Ohlson, J.M. Fredriksson, et al., J. Biol. Chem. 275 (2000) 25073-25081. DOI:10.1074/jbc.M000547200 |
| [28] |
Y. Mao, M. Zhang, J. Yang, et al., Toxicol. Res. 6 (2017) 297-304. DOI:10.1039/C6TX00410E |
| [29] |
J. Kreiter, A. Rupprecht, L. Zimmermann, et al., Biophys. J. 117 (2019) 1845-1857. DOI:10.1016/j.bpj.2019.10.021 |
| [30] |
M. Di Paola, M. Lorusso, Biochim. Biophys. Acta 1757 (2006) 1330-1337. DOI:10.1016/j.bbabio.2006.03.024 |
| [31] |
Z.G. Amerkhanov, I.Y. Kashapova, V.N. Popov, Dokl. Biochem. Biophys. 423 (2008) 328-330. DOI:10.1134/S1607672908060021 |
| [32] |
X.X. Yu, J.L. Barger, B.B. Boyer, et al., Am. J. Physiol. Endocrinol. Metab. 279 (2000) E433-E446. DOI:10.1152/ajpendo.2000.279.2.E433 |
| [33] |
D. Chretien, P. Benit, H.H. Ha, et al., PLoS Biol. 16 (2018) e2003992. DOI:10.1371/journal.pbio.2003992 |
| [34] |
K. Okabe, N. Inada, C. Gota, et al., Nat. Commun. 3 (2012) 705. DOI:10.1038/ncomms1714 |
| [35] |
Y. Shang, Y. Liu, L. Du, et al., Hepatology 50 (2009) 1204-1216. DOI:10.1002/hep.23121 |
| [36] |
C. Beall, K. Piipari, H. Al-Qassab, et al., Biochem. J. 429 (2010) 323-333. DOI:10.1042/BJ20100231 |
| [37] |
F. Shen, W. Yang, J. Cui, et al., Anal. Chem. 93 (2021) 13417-13420. DOI:10.1021/acs.analchem.1c03554 |
| [38] |
W. Liu, Z. Li, S. Chu, et al., Acta Pharm. Sin. B 12 (2022) 135-148. DOI:10.1016/j.apsb.2021.09.013 |
| [39] |
L. Contreras, E. Rial, S. Cerdan, J. Satrustegui, Neurochem. Res. 42 (2017) 108-114. DOI:10.1007/s11064-016-1999-5 |
| [40] |
D.A. Shannon, E. Weerapana, Curr. Opin. Chem. Biol. 24 (2015) 18-26. DOI:10.1016/j.cbpa.2014.10.021 |
| [41] |
K.D. Li, K. Yan, Q.S. Wang, et al., Food Funct. 10 (2019) 4533-4545. DOI:10.1039/c9fo00480g |
| [42] |
Y. Li, H. Pan, X. Li, et al., Toxicol. Appl. Pharmacol. 377 (2019) 114624. DOI:10.1016/j.taap.2019.114624 |
| [43] |
Y. Gao, J. Xu, G. Liu, et al., Materials 14 (2021) 6594. DOI:10.3390/ma14216594 |
| [44] |
M. Gehringer, S.A. Laufer, J. Med. Chem. 62 (2019) 5673-5724. DOI:10.1021/acs.jmedchem.8b01153 |
| [45] |
W.A. Denny, Curr. Med. Chem. 8 (2001) 533-544. DOI:10.2174/0929867003373283 |
| [46] |
C. Liu, F. Hao, J. Hu, et al., J. Proteome Res. 9 (2010) 6774-6785. DOI:10.1021/pr100970q |
| [47] |
G. Platzer, M. Okon, L.P. McIntosh, J. Biomol. NMR 60 (2014) 109-129. DOI:10.1007/s10858-014-9862-y |
| [48] |
T.K. Harris, G.J. Turner, IUBMB Life 53 (2002) 85-98. DOI:10.1080/15216540211468 |