 2023, Vol. 34
2023, Vol. 34 


b Key Laboratory of Precise Synthesis of Functional Molecules of Zhejiang Province, Department of Chemistry, School of Science and Research Center for Industries of the Future, Westlake University, and Westlake Institute for Advanced Study, Hangzhou 310030, China
As two of the most prevalent and well-known heterocycles, coumarin, and furan rings share some similarities [1-4], for example, they are both essential and abundant in biologically active natural products as well as various synthetic materials, and both have a diversity of biological and pharmaceutical properties. Moreover, both of them are extremely important building blocks and key structural elements in synthetic organic chemistry and medicinal chemistry.
When coumarin and furan rings are fused reciprocally, a unique and valuable family of heterocyclic compounds known as furocoumarins is formed (Fig. 1) [5-9]. The incorporation of coumarin and furan fragments into one polycyclic framework renders furocoumarins highly aromatic skeleton, extensively conjugated chromophores, and diverse functional sites, which may confer them synergistic biological activity, promising or even unprecedented pharmaceutical properties, and enhanced performance in various applications [8-14].

|
Download:
|
| Fig. 1. The furocoumarin family and its members. | |
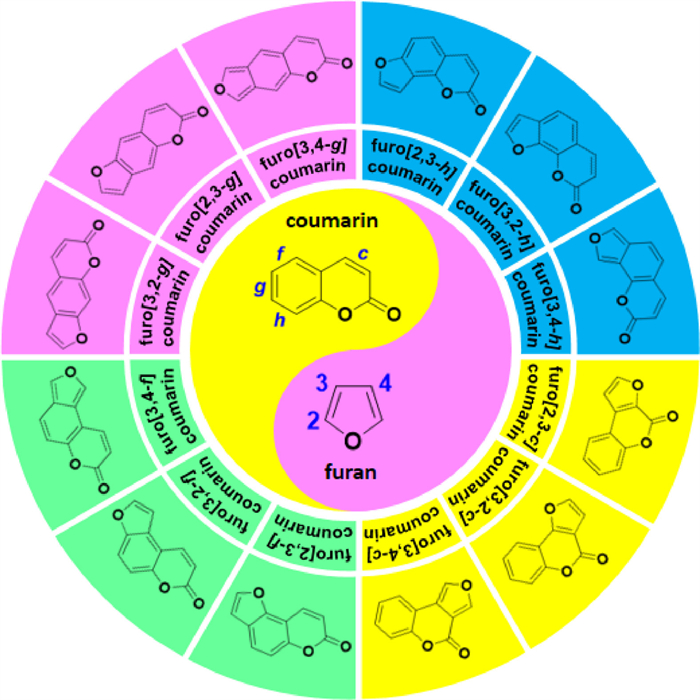{kind=link}
Indeed, furocoumarins have been frequently identified in the literature as one of the most active pharmacophores in medicinal chemistry during the last few decades [5-9]. They exhibit diverse and significant biological and pharmacological properties, such as anticancer, antivirals, antifungal, antimicrobial, antioxidant, anti-Alzheimer, anti-inflammatory, anti-depressant, HIV inhibition as well as antidiabetic treatment [15-22]. Other than their medicinal properties, they are also found in other important applications like chemosensors, photosensitizers, fluorescent probes, etc. [21,22].
Theoretically, the coumarin unit's c, f, g, or h bonds can be fused with the 2,3-, 3,2- or 3,4-position of the furan ring, respectively, resulting in the formation of numerous linear or angular isomeric derivatives of furocoumarins (Fig. 1) [8-10],17,20]. Within the furocoumarin family, some members such as psoralens, angelicins, allopsoralens, and coumestans are the most popular and abundant, whereas others are seldom explored and still far from practical.
The consistently growing trend of publications concerning furocoumarin derivatives for biological and pharmacological applications over the last several years demonstrates the vivid research interest in this topic. The purpose of this review is to summarize the most recent methods reported for the synthesis of the furocoumarin family, along with their applications in medicinal chemistry. Due to the limited space, this review will only highlight the advances over the past 5 years, and furocoumarins isolated from natural sources are considered outside the scope of this review.
2. Recent advances in furocoumarins synthesisEarlier related reviews have summarized the different synthetic methods by reaction types [7,9], starting materials [11], or type of newly formed ring in the transformations [5,6,8]. In this review, we divide the synthetic methods summary section into four parts according to the type of furocoumarin isomers synthesized: furo[g]coumarin, furo[c]coumarin, furo[f]coumarin, and furo[h]coumarin, respectively. Other transformations [23-34] in which the furocoumarin skeleton already exists in the molecule are not described here.
2.1. Synthesis of furo[g]coumarinsThe linear isomers in the furocoumarin family, furo[2,3-g], furo[3,2-g], and furo[3,4-g]coumarins, are also known as psoralens. They represent the most synthesized and intensely explored class of furocoumarins with widespread applications [5-9],18].
Although numerous new linear furo[g]coumarins have been synthesized in the last five years, the vast majority of them were prepared using the most classic and well-established methods to construct the tricyclic nucleus. Usually, these synthetic routes involve the initial Williamson condensation of hydroxycoumarin with an α-halo ketone (or α-halo aldehyde) to obtain the keto ether of coumarin, followed by subsequent intramolecular cyclization affording various functionalized furo[g]coumarin derivatives [35-43]. The wide scope of the starting materials, the cheapness of the catalysts, and mild reaction conditions keep this traditional approach appealing to many researchers even today. Since earlier reviews have introduced this synthetic route in detail, we will not comment on it further here.
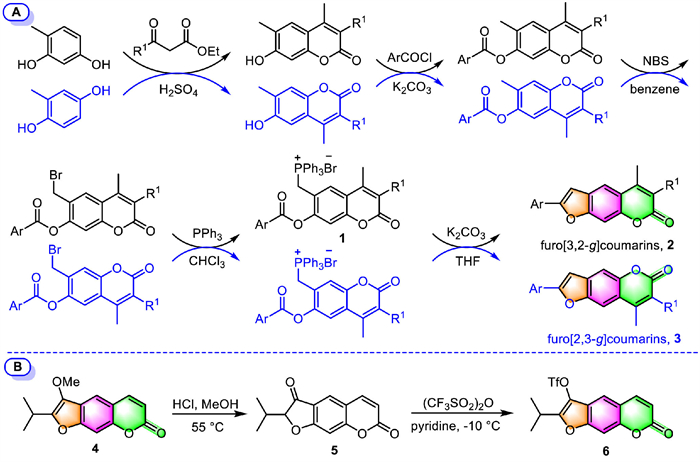
Apart from the above, a different synthesis route has been developed in 2021 by Luo's group for the production of a series of 2-aryl furocoumarins (furo[3,2-g]coumarins, 2) by employing an intramolecular Wittig reaction as the key step [44]. The synthesis involves in situ generations of the phosphorus ylides 1 via Michael addition reaction, followed by an intramolecular Wittig reaction in the presence of K2CO3 to finally annulate the furan ring onto the coumarin scaffold (Scheme 1A). Since the crude products in bromination and Michael addition steps are both humidity-sensitive substances, which were directly used in the next reaction without further purification. Using a similar synthetic route and reaction conditions, the same group also successfully synthesized a series of neofurocoumarin isomers (furo[2,3-g]coumarins, 3) by simply replacing the starting material 4-methylresorcinol with 2-methylbenzene-1,4-diol [45].

|
Download:
|
| Scheme 1. Synthesis of furo[3,2-g]coumarin/furo[2,3-g]coumarin via an intramolecular Wittig reaction (A) or a reversible dearomatization/aromatization process (B). | |
{kind=link}
Kremis et al. reported a synthetic approach to obtain 2,3-disubstituted psoralens based on the reversible dearomatization/aromatization process of the furan ring in tricyclic coumarins, respectively in 2019 [46] and 2022 [47]. Peucedanin (4), a natural furocoumarin isolated from Peucedanum morisonii roots, was hydrolyzed with the elimination of the methyl group in the presence of HCl, giving dearomatized 2,3-dihydrofurocoumarin (oreoselone, 5) quantitatively (Scheme 1B). The subsequent aromatization of the 2,3-dihydrofurocoumarin 5 by trifluoromethane sulfonic anhydride in the presence of pyridine afforded 2,3-disubstituted psoralens 6 in up to 72% yield.
2.2. Synthesis of furo[c]coumarinsWhen the furan ring is fused with the α-pyrone moiety of coumarin in different manners, three angular furocoumarin isomers, furo[2,3-c], furo[3,2-c], and furo[3,4-c] coumarin, are formed [48]. The literature survey revealed that among all the studies on furocoumarin derivatives synthesis, furo[c]coumarins are the class of the most actively researched and developed among the whole furanocoumarin family, with many novel methods for the synthesis of such compounds discovered in the last 5 years.
2.2.1. Furo[3,2-c]coumarinsDespite the diversity of methods used to synthesize furo[3,2-c]coumarins, the majority of them have one thing in common: The key building block is typically the inexpensive, readily available, and easily handled 4-hydroxycoumarins. This is due to the presence of numerous active reaction sites in 4-hydroxycoumarins, including electrical centers at the C-2 and C-4 positions, as well as nucleophilic centers at the C-3 position and the oxygen of the 4-OH group [49,50]. These reaction sites enable 4-hydroxycoumarins to perform a number of transformations with various reaction partners in order to form a new furan ring on the pyranone ring. For clarity, we divide these methods into two-component reactions, multi-component reactions (MCRs), and so on, and discuss them separately.
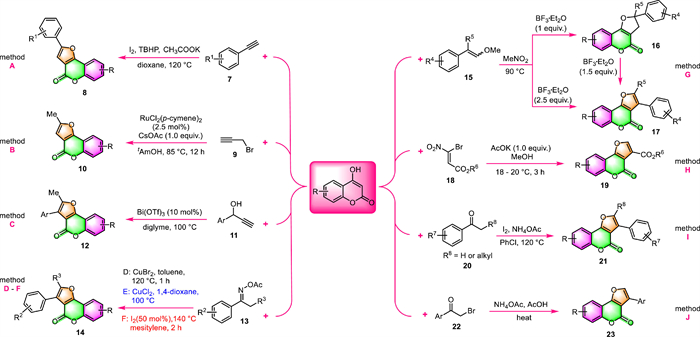
First, 4-hydroxycoumarin derivatives and alkynes are suitable cross-coupling partners for constructing furo[3,2-c]coumarins via a [3 + 2] cycloaddition strategy. In 2018, Chu and co-workers devised an operationally simple and efficient synthesis of furo[3,2-c]coumarins 8 through I2/TBHP-mediated cross-coupling of terminal alkynes 7 with 4-hydroxycoumarins under aerobic conditions (Scheme 2, method A) [51]. The use of molecular iodine I2 as the catalyst makes this protocol a convenient, cost-effective, nontoxic, and environmental-friendly method for the synthesis of furo[3,2-c]coumarins 8.

|
Download:
|
| Scheme 2. Synthesis of furo[3,2-c]coumarins by various two-component reactions using 4-hydroxycoumarins as the raw materials. | |
{kind=link}
The group of Gogoi disclosed the use of propargyl bromide 9 as a partner in the debrominative coupling reaction with 4-hydroxycoumarin, to provide methyl-substituted furo[3,2-c]coumarin 10 [52]. This one-pot reaction was promoted by [RuCl2(p-cymene)]2 in the presence of CsOAc in tAmOH, and the yield of the product reached 86% (Scheme 2, method B).
Following the above pioneering studies, Kim and co-workers expanded the substrate scope to include terminal propargyl alcohols 11 by using Bi(OTf)3 as the catalyst and diglyme as the solvent [53]. The corresponding furo[3,2-c]coumarins 12 were smoothly obtained via initial propargylation of 4-hydroxycoumarins followed by intramolecular cyclization of the resulting propargylated coumarins (Scheme 2, method C). It is noteworthy that Bi(OTf)3 plays bifunctional roles in effectively catalyzing these two reactions in one pot.
As a protected form and also a typical precursor of ketones, ketoxime acetates 13 have been proven to be adaptable coupling partners with 4-hydroxycoumarins to generate furocoumarins. During those transformations, the unstable N–O bond in ketoxime acetates 13 was easily cleaved to generate active N-centered iminyl radicals via a single-electron transfer (SET) pathway, thus triggering the formation of furo[3,2-c]coumarins.
For example, Phan's group performed a CuBr2-catalyzed cyclization between ketoxime acetates 13 and 4-hydroxycoumarins to produce substituted furocoumarins 14, in which ketoxime acetates acted as both a reactant and an internal oxidant (Scheme 2, method D) [54]. This synthetic strategy features cheap catalyst CuBr2, readily available starting materials, and avoidance of the utilization of stoichiometric oxidants.
Almost at the same time, He and co-workers independently described CuCl2-catalyzed synthesis of substituted furo[3,2-c]-coumarins 14 with the same substrates (Scheme 2, method E) [55]. This protocol affords a range of structurally diverse furocoumarins, including 2-substituted, 3-substituted, 2,3-disubstituted, and 2,3-fused ones, via a radical/radical cross-coupling process.
In 2020, Pham et al. carried out the same transformations in a metal-free fashion and under mild and simple conditions (Scheme 2, method F) [56]. They introduced eco-friendly iodine as a catalyst to activate the N–O bond of ketoxime acetates 13 for subsequent formal [3 + 2] annulation of 4-hydroxycoumarin in mesitylene to synthesize furocoumarins 14. In addition, this protocol has broad substrate scope and good tolerance of functional groups, even steric bulky ketoxime acetates could also be successfully transformed into the corresponding products.
Very recently, the group Khan has described a one-pot, BF3·Et2O-controlled, and mild protocol for the selective synthesis of furo[3,2-c]coumarin derivatives from 4-hydroxycoumarin and methyl enol ethers (MEE, 15) (Scheme 2, method G) [57]. Moreover, this procedure could easily be scaled up to gram-scale synthesis using the standard protocol. The most important feature of this approach is that dihydrofuro[3,2-c]chromenones 16 and furo[3,2-c]coumarins 17 could be selectively obtained by carefully controlling the stoichiometry of BF3·Et2O. The mechanism investigation revealed that the furo[3,2-c]coumarins derivatives were constructed via an interesting aryl group migration followed by the aromatization of furan moiety.
In 2022, Pelipko and colleagues prepared two furo[3,2-c]coumarin-3-carboxylates 19 in anhydrous MeOH via a straightforward reaction of 4-hydroxycoumarin with accessible alkyl 3–bromo-3-nitroacrylates 18 in the presence of 1.0 equiv. AcOK (Scheme 2, method H) [58]. The transformation was completed in 3 h under mild room temperature (18–20 ℃), with yields of 77% and 80%.
Pham and co-workers obtained furocoumarins 21 via an iodine-promoted one-pot cyclization of 4-hydroxycoumarins with acetophenones 20 (Scheme 2, method I) [59]. NH4OAc was found to be the best additive in this protocol, while both the acidic and basic additives failed to improve the reaction yields. In addition, the solvent played a crucial role in this transformation, and chlorobenzene proved to be the best choice of solvent. Mechanism investigation suggested that the reaction would proceed by a 5-exo-tet cyclization rather than an O-alkylation. Notable advantages of this protocol include the avoidance of transition-metal catalysts, readily available and inexpensive materials, excellent yields, and wide substrate scope.
Conventionally, furo[3,2-c]coumarin derivatives can be prepared through a well-established two-step procedure involving the first Williamson condensation of 4-hydroxycoumarins with α-halo ketones followed by intramolecular cyclization. It is a convenient and economical method in terms of cost and availability of starting materials. However, it also suffers from the drawbacks of multistep protocols and troublesome work-up procedures. Patel's group simplified the two-step processes into a one-pot fashion under metal-free conditions in 2020 (Scheme 2, method J) [60]. Various 3-aryl-furo[3,2-c]coumarins 23 were achieved in moderate to good yields in the presence of NH4OAc in refluxing acetic acid. The authors proposed that the reaction proceeds through the Michael addition of the active methylene function of 4-hydroxycoumarin with α–bromo ketones 22.
Subsequent to the work of Patel [60], Rani and colleagues disclosed that 2,3-disubstituted furo[3,2-c]coumarins may be produced in one step by using 3-acyl substituted 4-hydroxycoumarins (24, 25) instead of 4-hydroxycoumarins to react with α–bromo ketones [61]. The target furo[3,2-c]coumarin derivatives (26 and 27) were obtained from 3-acetyl-4-hydroxycoumarin 24 or its chalcone 25 by reaction with various α–bromo ketones in refluxing acetonitrile in the presence of K2CO3 (Scheme 3). The distinction is that the products are generated via the Williamson reaction/intramolecular aldol condensation pathway rather than the Michael addition method.

|
Download:
|
| Scheme 3. Synthesis of furo[3,2-c]coumarins by reaction of 3-acetyl-4-hydroxycoumarin and its chalcone with α–bromo ketones. | |
{kind=link}
As a powerful and versatile methodology, MCRs (usually three-component reactions) have found increasing application in the synthesis of furocoumarins [5-7]. Employing 4-hydroxycoumarins as one of the raw materials, many novel furo[3,2-c]coumarins were synthesized through three-component reactions in a one-pot fashion.
Formerly, Nair and co-workers described an impressive synthetic protocol to access furocoumarin derivatives through the one-pot three-component condensation of 4-hydroxycoumarin, aldehyde, and cyclohexyl isocyanide in 2002 [62]. This reaction is performed under simple reflux conditions in benzene, and most presumably occurs via a sequential [4 + 1]-cycloaddition followed by a [1,3]-H shift process. The simplicity of this procedure without using any catalyst or additive makes it an interesting alternative to other approaches.
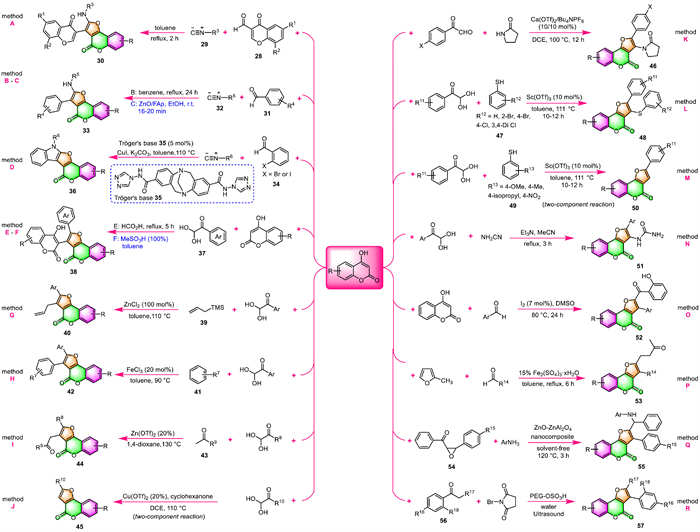
In 2019, the substrate scopes of aromatic aldehydes and isocyanides in this MCR were further extended to respective 3-formylchromones 28 and alkyl or aryl isocyanides 29 by Meydani and co-workers (Scheme 4, method A) [63]. A series of furo[3,2-c]coumarin-chromone conjugates 30 were achieved in refluxing toluene in only 2 h. They proposed a similar mechanism to that reported by Nair and co-workers involving sequential Knoevenagel condensation, [4 + 1]-cycloaddition, and imine-enamine tautomerization [1,3]-H shift.

|
Download:
|
| Scheme 4. Synthesis of furo[3,2-c]coumarins by various MCRs using 4-hydroxycoumarins as the raw materials. | |
{kind=link}
Following the same MCR procedure reported by Nair and co-workers, Maher's group [64,65] and Idris's group [66] have respectively prepared some furo[3,2-c]coumarin derivatives 33 with excellent fluorescence properties starting from 4-hydroxycoumarin, cyclohexyl isocyanide 32, and aromatic aldehydes 31 (Scheme 4, method B).
Jonnalagadda's group has transformed the above MCR into a greener and more sustainable approach by using zinc oxide loaded on fluorapatite (ZnO/FAp) as a heterogeneous catalyst (Scheme 4, method C) [67]. Ten new furo[3,2-c]coumarins 33 were very efficiently obtained with excellent yields (94%–98%) within 20 min at room temperature in EtOH. ZnO/FAp catalyst could be recycled and reused up to 5 catalytic cycles without appreciable loss of its catalytic activity. The mild reaction conditions, excellent yields, reusability of the catalyst, and operational simplicity together with the avoidance of column chromatography make the present procedure sustainable and advantageous compared to existing protocols.
When ortho-halo benzaldehydes 34 were used as one of the reactants in this MCR, Zhou and coworkers [68] confirmed that indole-fused furocoumarins 36 with a pentacyclic skeleton could be synthesized under the co-catalysis of Tröger's base derivative 35 and CuI (Scheme 4, method D). The 1H NMR titration and control experiments showed that those indole-fused furocoumarins 36 were formed through cascade Aldol-[4 + 1]cycloaddition-intramolecular Ullmann reaction.
In 2018, Gorbunov and co-workers reported a one-pot synthesis of furocoumarin-coumarin conjugates 38 through the multicomponent condensation of two equivalents of 4-hydroxycoumarin with various arylglyoxals 37 in refluxing formic acid (Scheme 4, method E) [69]. The target products 38 were produced in yields from 42% to 72%, and they could be isolated simply by recrystallization from ethanol.
Very shortly afterward, Chen's group conducted the same MCR using MeSO3H as the catalyst instead of formic acid (Scheme 4, method F [70]). This protocol produced the expected furocoumarin-coumarin conjugates 38 with relatively higher yields than previously reported. Moreover, this procedure displays a broad substituent scope, arylglyoxals 37 bearing electron-neutral, electron-rich, and electron-deficient substituents along with halo-substituted were reacted smoothly with 4-hydroxycoumarins to afford the corresponding products.
Encouraged by the above work, the same authors later developed several closely related approaches to synthesize furo[3,2-c]coumarin derivatives, which are based on Lewis acid-mediated multicomponent tandem reactions. In 2019 [71], they reported the three-component assembling of 4-hydroxycoumarin, arylglyoxal, and allyl trimethyl silane 39 in the presence of ZnCl2 as the Lewis acid catalyst at 110 ℃ in toluene. As a result, they obtained a variety of 3-allyl furo[3,2-c]coumarins 40 in moderate yields (Scheme 4, method G). Additionally, when substituted benzenes 41 were employed as one of the substrates to replace allyl trimethyl silanes 39 in this protocol, and using FeCl3 as the catalyst, the corresponding 3-aryl furo[3,2-c]coumarins 42 were obtained instead in 48%–82% (Scheme 4, method H).
Again in 2021, they further demonstrated that the prescribed Lewis acid-mediated MCRs could be applied to a wide variety of ketone nucleophiles 43 to provide furocoumarin analogs with necessary modification [72]. As shown in Scheme 4 (method I), the three-component reactions were carried out in 1,4-dioxane at 130 ℃ in the presence of Zn(OTf)2, furnishing 3-acylmethyl furo[3,2-c]coumarins 44 in moderate to good yields. The Brønsted acids such as p-toluenesulfonic acid (TsOH) and MeSO3H were proved less effective as catalysts than Lewis acids. However, aliphatic cyclic ketones failed to give the desired three-component products under standard conditions. On the contrary, two-component products of 3-unsubstituted furo[3,2-c]coumarins 45 were obtained instead when cyclohexanone (2.0 equiv.) was heated with 4-hydroxycoumarin and various arylglyoxals in 5 mL of DCE at 110 ℃ in a sealed vessel under the catalysis of Cu(OTf)2 (Scheme 4, method J).
Later in 2021, Yaragorla's group fulfilled a similar transformation by using 2-pyrrolidinone as one of the reactants with Ca(OTf)2 and Bu4NPF6 as the co-catalyst (Scheme 4, method K) [73]. The key intermediate exocyclic N-acyliminium ions (NAIs) were generated in situ and directly incorporated with 4-hydroxycoumarin, furnishing the furo[3,2-c]coumarin derivatives 46 in 50%–56% yields.
Very recently, Choudhury's group has presented an efficient one-pot methodology for the construction of furocoumarins via a three-component reaction of 4-hydroxycoumarins, arylglyoxal, and various thiols (47 and 49) (Scheme 4, method L and M) [74]. In this protocol 10 mol% Sc(OTf)3 was utilized for the catalyst and toluene served as the solvent. It is of interest that this method leads to two different products depending upon the types of substituents present in the aryl thiols. Under the standard reaction conditions, aryl thiols 47 bearing 4-H, 4-Cl, 4-Br, or 2-Br groups afforded three-component thioether-linked furocoumarins 48 as the major products (method L), while those bearing 4-OMe, 4-Me, 4-isopropyl, 4-nitro groups (49) provided two-component products 50 instead (method M). The merits of this methodology include broad substrate scope, good to excellent yields, and products containing multiple pharmaceutically important units.
In 2022, Komogortsev and co-workers [75] have synthesized various urea-substituted furocoumarins 51 by multicomponent condensation of 4-hydroxycoumarins, arylglyoxals, and cyanamide, using Et3N as the catalyst (Scheme 4, method N). The work-up procedure is very simple, and the pure products can be obtained simply by recrystallization in good yields.
By following the earlier reported method of Kolita et al. [76] with a slight modification, Fattah and co-workers have prepared a series of functionalized furo[3,2-c]coumarins 52 via a one-pot three-component tandem approach (Scheme 4, method O) [77]. The reaction occurred through condensation of 4-hydroxycoumarins with aldehydes/aryl methyl ketones mediated by iodine in DMSO. In this reaction process, DMSO acts as both solvent and mild oxidizing agent and also recycles the iodine in situ.
A Fe-catalyzed three-component assembly of 4-hydroxycoumarin, 2-methylfuran, and aryl- or alkyl-aldehyde has been developed by Noland and co-workers in 2020, providing various 2,3-disubstituted furo[3,2-c]coumarins 53 (Scheme 4, method P) [78]. Fe2(SO4)3·xH2O, the most efficient, cheap, and readily available Lewis acid, was selected as a prominent catalyst for this reaction. In addition, this protocol can be readily scaled-up to the gram level.
Ghashang's group also developed a one-pot three-component process for the synthesis of 2,3-disubstituted furo[3,2-c]coumarins 55 from 4-hydroxycoumarin, aromatic amines, and α,β-epoxy ketones 54, using ZnO-ZnAl2O4 nanocomposite as the catalyst (Scheme 4, method Q) [79]. It is worth noting that this reaction was carried out under solvent-free conditions. In particular, the ZnO-ZnAl2O4 nanocomposite catalyst used in this process could be easily recycled and reused at least five times without an obvious decrease in catalytic performance.
Wagare and co-workers developed a one-pot, multi-components, and eco-friendly approach for the synthesis of furo[3,2-c]coumarins [80]. A series of biologically active 3-aryl furo[3,2-c]coumarins 57 were efficiently obtained by cyclocondensation of 4-hydroxycoumarins with acetophenones 56 and N–bromo succinimide (NBS) using PEG-OSO3H in water under ultrasonic promotion at 300-watt power (Scheme 4, method R). The features of this reaction include using water as the solvent, reusability of the PEG-OSO3H catalyst, utilization of ultrasound radiation, and in situ generation of toxic lachrymators and unstable phenacyl bromides.
There are also examples of three-component reactions that produce furo[3,2-c]coumarins in a one-pot, two-stage process.
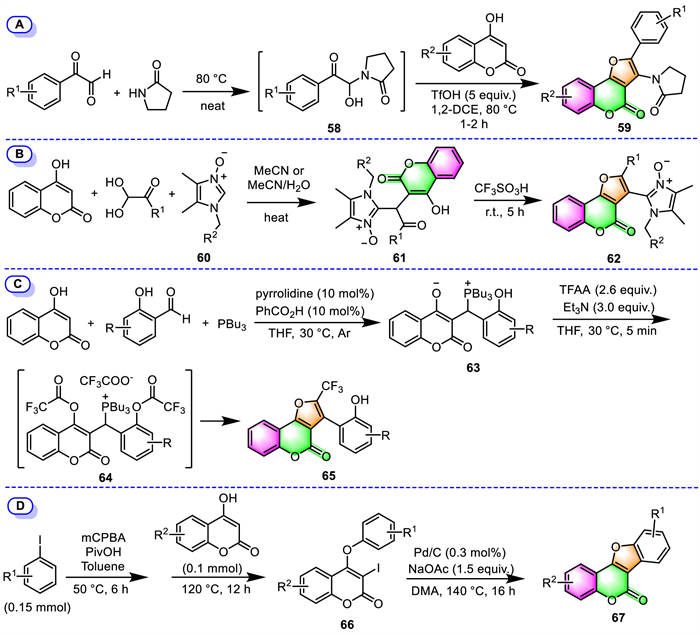
For example, different from the process [73] reported by Yaragorla's group mentioned above (Scheme 4, method K), Sharada's group performed the same MCR of 4-hydroxycoumarins, arylglyoxals, and 2-pyrrolidinone in a one-pot, two-stage manner to prepare fully substituted furo[3,2-c]coumarins 59 (Scheme 5A) [81]. In this transformation, the same key precursors NAIs 58 were firstly generated in situ from arylglyoxals and 2-pyrrolidinone under catalyst- and solvent-free conditions, followed by a triflic acid-promoted tandem cyclization with 4-hydroxycoumarins in the same pot, delivering eight furocoumarins 59 in good to excellent yields.

|
Download:
|
| Scheme 5. Synthesis of furo[3,2-c]coumarins via one-pot, two-stage route using 4-hydroxycoumarins as one of the raw materials. | |
{kind=link}
In 2020, Kutasevich's group has accessed two imidazole-substituted furo[3,2-c]coumarins 62 through a two-step process involving the initial three-component condensation of 4-hydroxycoumarin, arylglyoxals with imidazole N-oxides 60, and subsequent CF3SO3H-promoted cyclization of the resulting 3-substituted-4-hydroxycoumarins 61 at ambient temperature (Scheme 5B) [82]. This two-step process is easy to work up, the reaction intermediates 61 and target products 62 were obtained by simple recrystallization without column chromatography purification.
Phosphorus ylides are multipurpose and readily accessible intermediates in organic synthesis. Lin’ group has demonstrated that the phosphorus ylide intermediates, which were prepared by three-component condensation of 4-hydroxycoumarin and 2-hydroxybenzaldehydes with PBu3, can serve as efficient building blocks for the construction of furo[3,2-c]coumarins (Scheme 5C) [83]. In 2018, they prepared various 3-aryl-2-trifluoromethyl furo[3,2-c]coumarins 65 via chemoselective acylation/Wittig reaction starting from their novel phosphorus ylides with commercially available TFAA. The process proceeds in a one-pot fashion: subjecting phosphorus ylides to reaction with TFAA and Et3N in dry THF under an argon atmosphere, leading to in situ formation of the bisacylated phosphonium salts 64, followed by chemoselective intramolecular Wittig reaction to provide furo[3,2-c]coumarins 65.
Panda and co-workers have described the high-yield preparation of tetracyclic furo[3,2-c]coumarins derivatives (coumestans, 67) by intramolecular annulation of 3-iodo-4-aryloxy coumarins 66 through C–H activation using commercially viable Pd/C catalyst under ligand-free conditions (Scheme 5D) [84]. Their synthetic precursors are easily accessible by sequential iodination and O-arylation of 4-hydroxycoumarins with aryl iodides in a one-pot two-step process. The important features of this reaction include using ligand-free conditions, a simple work-up procedure, and the recoverability and reusability of the palladium catalyst.
In addition to the methods described above, there are a few alternative ways to synthesize furo[3,2-c]coumarins that do not use 4-hydroxycoumarin as the basic material.
By using sustainable CO2 as a C1 source, Fu and co-workers developed a novel method to construct pyranone rings through rhodium(Ⅱ)-catalyzed aryl C–H carboxylation of 2-furanylphenols 68 (Scheme 6A) [85]. The carboxylation procedure was carried out for 48 h at ambient CO2 pressure using diglyme as the solvent and Rh2(OAc)4 and PCy3 as the ligands. Seven furano[3,2-c]coumarin derivatives 69 were smoothly produced in 70%–86% yields.

|
Download:
|
| Scheme 6. Some methods for synthesizing furo[3,2-c]coumarins without using 4-hydroxycoumarin as the basic material. | |
{kind=link}
Vagh and his co-workers have developed two efficient one-pot reactions to generate functionalized furo[3,2-c]coumarins 71 and 2,3-disubstituted furo[3,2-c]coumarins 72, both employing alkynoates 70, PR3, and acyl chlorides as starting materials (Scheme 6B) [86]. Thus, through a one-pot MBH-type/acyl-transfer/Wittig reaction of terminal alkynoates 70 with acyl chlorides, mediated by phosphine in the presence of DIPEA, a series of functionalized furo[3,2-c]coumarins 71 were obtained in moderate to high yields. It is noteworthy that the pyrone ring and furan ring are simultaneously formed in one pot, and at the same time, keto functionality is directly embedded into the furan ring of furo[3,2-c]coumarin via an unprecedented acyl-transfer process under metal-free conditions.
When terminal alkynoates 70 were replaced by internal ones, another one-pot approach through a domino sequence of MBH-type and intramolecular Wittig reaction was established, and 2,3-disubstituted furo[3,2-c]coumarins 72 were achieved under the same reaction conditions.
Yu's group has conducted a series of studies on the synthesis and structure-activity relationship of coumestan derivatives [87-90], a kind of naturally occurring tetracyclic furocoumarins. They employed privileged 2-(2-methoxyphenyl)benzofuran derivatives 73 as the key intermediates to construct the corresponding coumestan framework 74 (Scheme 6C). The conversion was performed in one pot by the first demethylation using BBr3 and subsequent intramolecular lactonization/transesterification in refluxing ethanol [89,90]. The key intermediates, 2-(2-methoxyphenyl)benzofuran derivatives 73, can be facially prepared through Cu(OTf)2-catalyzed cross-dehydrogenative coupling (CDC) of 1,4-benzoquinones with substituted ethyl 2-benzoylacetate.
A similar dealkylation/intramolecular lactonization strategy was also applied to the synthesis of a pentacyclic coumestan analog from a benzofuran derivative by Zhang and co-workers [91]. Firstly, 2-arylbenzofuran-3-carbaldehyde 75, which was prepared by an organocatalytic [3 + 2] annulation/oxidative aromatization reaction, was oxidized to the corresponding carboxylic acid 76 by NaClO2. Dealkylation of the resulting 2-arylbenzofuran-3-carboxylic acid 76 with Pd/C under H2 atmosphere and then lactonization by using TsOH·H2O furnishing the desired coumestan derivative 77 in moderate yield (Scheme 6D).
In addition to the most used 4-hydroxycoumarins with the –OH groups at the 4-position, 3-(2′-hydroxyaryl)coumarins 78, which have their –OH groups at the ortho-position of 3-aryl substituents, can also be employed as starting materials for the synthesis of furo[3,2-c]coumarins. In 2019, Song and co-workers disclosed an approach to afford coumestan derivatives 79 through a Cu(OAc)2-catalyzed intramolecular cross dehydrogenative C–O coupling of 3-(2′-hydroxyaryl)coumarins 78 (Scheme 6E, method a) [92]. This protocol exhibits high efficiency, moderate to high yields, and good functional group tolerance for phenolic hydroxyl groups. Moreover, the starting materials, 3-(2′-hydroxyaryl)coumarins 78, could be readily accessed through Perkin condensation of easily available 2–hydroxy phenylacetic acids with ortho-hydroxybenzaldehydes.
Also employing 3-(2′-hydroxyaryl)coumarins 78 as key intermediates, Shinde et al. disclosed an alternative protocol for the synthesis of coumestan derivatives 79 by a DDQ-mediated oxidative cyclization in toluene (Scheme 6E, method b) [93]. They also provided another synthetic option for the preparation of the 3-(2′-hydroxyaryl)coumarin intermediates 78, in which 3-arylcoumarins were efficiently ortho-hydroxylated using Pd(OAc)2 (10 mol%) as the catalyst, K2S2O8 (2 equiv.) as an oxidant, and TFA as an oxygen source.
Very recently, Zou's group performed the same transformation in H2O under air using 1,8-diazabicyclo[5.4.0]undec–7-ene (DBU) as the catalyst (Scheme 6E, method c) [94]. They proposed that the tandem reaction occurred via a sequential intramolecular dehydrogenation/oxa-Micheal reaction. This method has also been used in the total syntheses of three natural products, namely, 4′-O-methylcoumestrol, coumestrol, and plicadin.
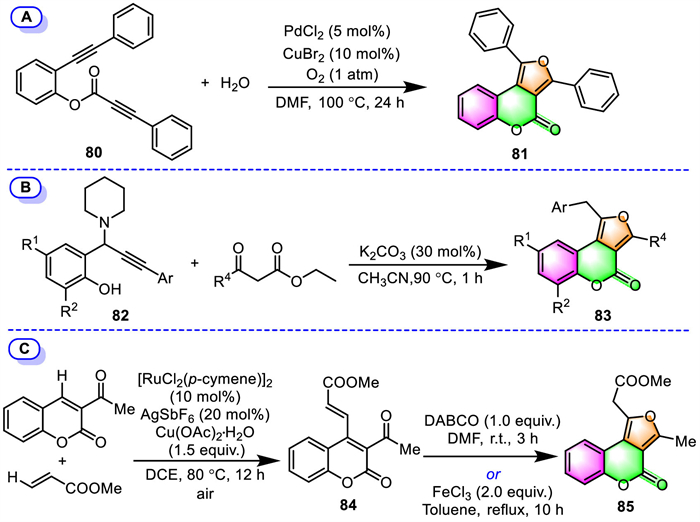
2.2.2. Furo[3,4-c]coumarinsOuyang and co-workers have revealed a novel palladium-catalyzed oxidative [2 + 2 + 1] annulation strategy for the synthesis of furo[3,4-c]coumarin [95]. In the only example provided, a tandem cyclization was performed between phenol-linked 1,7-diyne 80 and water under a PdCl2/CuBr2 catalytic system, furnishing the diphenyl furo[3,4-c]coumarin 81 in 68% yield (Scheme 7A).

|
Download:
|
| Scheme 7. Various methods for the synthesis of furo[3,4-c]coumarins. | |
{kind=link}
Li and co-workers demonstrated that multi-functionalized furo[3,4-c]coumarins can be built in a regioselective manner through a K2CO3-mediated sequential 1,4-conjugate addition/intramolecular 5-exo-dig annulation of propargyl amines 82 with β-keto esters under transition-metal free conditions (Scheme 7B) [96]. The reaction tolerated a variety of electron-deficient, electron-rich, and halogen-substituted propargyl amines. Even crowded propargyl amine with the bulk tert–butyl groups at both the ortho- and para-position of the –OH group could also afford the corresponding product 83 smoothly.
Another attempt aiming at the regioselective synthesis of furo[3,4-c]coumarins has been presented by Zhao's group in 2020 [97]. They developed a two-step process employing 3-acetylcoumarin and methyl acrylate as the starting materials (Scheme 7C). The process involves the initial direct C-4 alkenylation of 3-acetylcoumarin with methyl acrylate catalyzed by a [RuCl2(p-cymene)]2/AgSbF6/Cu(OAc)2·H2O catalytic system under air atmosphere, followed by the sequential intramolecular cyclization of the resulting 4-alkenylated coumarin 84. Interestingly, both DABCO and FeCl3 can be employed as cyclization catalysts in the formation of furo[3,4-c]coumarin 85.
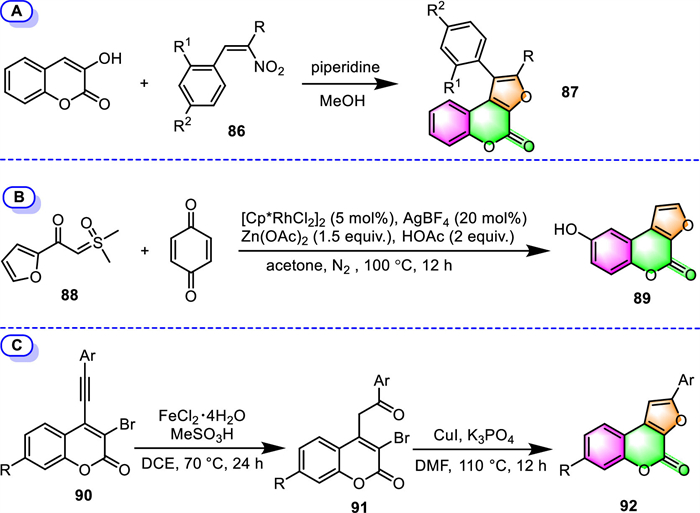
2.2.3. Furo[2,3-c]coumarinsIn the same way that 4-hydroxycoumarin is commonly used for the synthesis of furo[3,2-c]coumarins, 3-hydroxycoumarin can correspondingly be used as the building block for the synthesis of another furocoumarin family member, the furo[2,3-c]coumarin derivatives. Pandya and co-workers disclosed the use of 3-hydroxycoumarin and β-nitrostyrene derivatives 86 as the raw material to provide 1-phenyl-furano[2,3-c]coumarins (Scheme 8A) [98]. This one-pot reaction takes place under piperidine promotion in refluxing MeOH, and the 87 are achieved in yields of 55%–61%.

|
Download:
|
| Scheme 8. Various methods for the synthesis of furo[2,3-c]coumarins. | |
{kind=link}
Dong and co-workers recently reported the use of Rh(Ⅲ)-catalyzed cascade reaction to furnish 8–hydroxy-furo[2,3-c]coumarin 89 using sulfoxonium ylide 88 and DDQ as the reaction partners (Scheme 8B) [99]. This process begins with ortho-C–H oxidative functionalization of sulfoxonium ylide 88, followed by intramolecular annulation with DDQ. Unfortunately, the product 8–hydroxy-furo[2,3-c]coumarin 89 was obtained with only a 25% yield.
An efficient two-step synthesis of furo[2,3-c]coumarins has been recently developed by Rao's group employing 4-(arylethynyl)-3-bromocoumarins 90 as the starting materials (Scheme 8C [100]). The stepwise synthetic process involves the initial formation of 3–bromo-4-(2-oxo-2-arylethyl)coumarins 91, by means of catalytic FeCl2·4H2O, and a subsequent intramolecular cyclization catalyzed by CuI in the presence of potassium phosphate. A series of furo[2,3-c]coumarin derivatives 92 were obtained in moderate to high yields (up to 98% yield).
2.3. Synthesis of furo[h]coumarinsFuro[2,3-h]coumarins are also known as angelicins or isopsoralens. Of the many known synthesis methods of furo[2,3-h]coumarin derivatives, the most straightforward and often employed approaches are the annelation of a furan ring to coumarin by intramolecular cyclocondensation of 7-O-keto ether of coumarin or intermolecular cyclocondensation of 8-acyl-7–hydroxy coumarin with α-halo ketones. These well-established classical methods are still widely used with some necessary modifications.
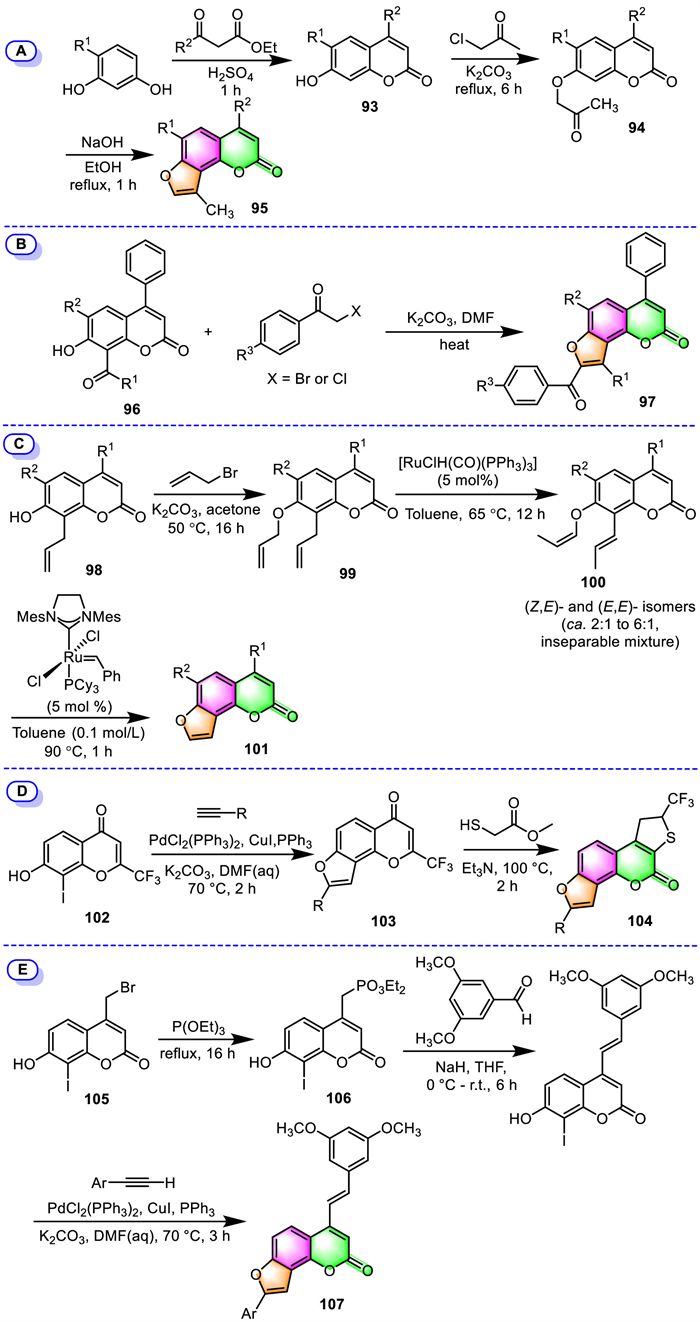
One of the examples is presented by Chilin and his colleagues [101], who prepared seven furo[2,3-h]coumarin derivatives 95 bearing sterically more hindered substituents at the 4-position, aiming for minimizing or avoiding the covalent photoreactions between substituted angelicins and DNA (Scheme 9A). They followed a classical synthetic route involving the initial condensation of 4-alkyl resorcinols with acetoacetic esters, and subsequent etherification of the resulting alkyl-7-hydroxycoumarins 93 with chloroacetone. In the final key step, the furan ring was incorporated into the coumarin core by dehydrative cyclization of the keto ether 94 of coumarin in KOH/EtOH. A quite similar angelicins formation route was also disclosed by the group of Barraja [102].

|
Download:
|
| Scheme 9. Various methods for the synthesis of furo[2,3-h]coumarins. | |
{kind=link}
Using the frequently employed approach, Shokol and their co-workers successfully synthesized a series of 4-alkyl-8-aroyl-9-arylangelicin derivatives [103], furo[2,3-h]neoflavones [104], and one 3-(benzothiazol-2-yl)-substituted furo[2,3-h]coumarin (Scheme 9B) [105]. The key step in those approaches all involved the cyclocondensation of 8-acyl-7–hydroxy coumarins 96 with substituted α-halo ketones under K2CO3/DMF condition, forming the furan fragment and thus leading to the formation furo[2,3-h]coumarins 97.
Starting from easily accessible 8-allylcoumarins 98, the group of Schmidt reported a three-step synthesis of furo[2,3-h]coumarins by using ring-closing olefin metathesis (RCM) reactions as one of the key steps [106]. As shown in Scheme 9C, 8-allylcoumarins 98 were first O-allylated with 3-bromopropene, and then the resulting allyl ethers 99 underwent Ru hydride-catalyzed double bond isomerization to provide (Z,E)- and (E,E)-enol ethers 100 as a pair of inseparable diastereoisomers. Finally, an intramolecular RCM under the Grubbs’ catalyst was carried out on the mixture of enol ethers to complete the synthesis of furo[2,3-h]coumarins 101 in excellent yields (89% to quantitative).
During the synthesis of trifluoromethyl-substituted furo[2,3-h]coumarin analogues, Mphahlele's group employed Pd(II)/Cu(I) co-catalyzed Sonogashira cross-coupling and heteroannulation of 7–hydroxy-8-iodo-2-(trifluoromethyl)chromen-4-one 102 with terminal acetylenes to assemble the key intermediates 2,5-dicarbo substituted 4H-furo[2,3-h]chromen-4-ones 103 [107]. The final 1,4-nucleophilic ring addition of the resulting intermediates 103 with methyl mercapto acetate catalyzed by Et3N afforded the desired trifluoromethyl-substituted furocoumarins 104 in moderate-to-high yields (Scheme 9D).
A convenient three-step synthesis of furo[2,3-h]coumarin-stilbene hybrids, starting from commercially available 4-(bromomethyl)-7-methoxycoumarin 105, has been accomplished by the group of Agbo in 2020 (Scheme 9E) [108]. The reaction pathway involves sequential Arbuzov-type demethylation, followed by Horner-Emmons olefination, and finally tandem Sonogashira cross-coupling and subsequent Cacchi-type cycloisomerization 106 using Pd/Cu dual catalysis. The desired 4-stilbene appended furocoumarin hybrids 107 were achieved in 60%–79% yields.
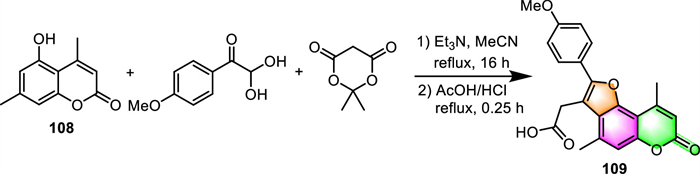
2.4. Synthesis of furo[f]coumarins 2.4.1. Furo[2,3-f]coumarinsIn 2021, Lichitsky and his associates established a one-pot, multi-component protocol for the synthesis of substituted furo[2,3-f]coumarin 109 via the reaction of 5–hydroxy-4,7-dimethyl-2H-chromen-2-one 108 with 4-methoxyphenylglyoxal and Meldrum's acid, using Et3N as a mild base (Scheme 10) [109]. The product can be easily obtained just by filtration instead of chromatographic separation. However, due to the low reactivity of 5–hydroxy coumarin, a prolonged reflux time (16 h) and a relatively high amount (6-fold excess) of arylglyoxal, Meldrum's acid, and Et3N are both needed to enhance the conversion of the starting materials.

|
Download:
|
| Scheme 10. Synthesis of furo[2,3-f]coumarin via condensation of 5–hydroxy-coumarin with phenylglyoxal and Meldrum's acid. | |
{kind=link}
2.4.2. Furo[3,2-f]coumarins
Singh and co-workers confirmed that salicylaldehyde derivative 110 is a versatile building block for the synthesis of furo[3,2-f]coumarins (Scheme 11) [110]. Upon catalyzing with piperidine, salicylaldehyde derivative 110 can undergo Perkin reaction with propionic anhydride to produce furocoumarin 111 in 46% yield, and Knoevenagel condensation with diethyl malonate to generate furocoumarin 112 in 62% yield, respectively.

|
Download:
|
| Scheme 11. Syntheses of furo[3,2-f]coumarins from salicylaldehyde derivative 110. | |
{kind=link}
Except for the developments summarized above, studies on the synthesis of the remaining five furocoumarin isomers have received extremely little attention from synthetic chemists and have not been reported in the last five years.
3. Biological and pharmaceutical activities of furocoumarin derivativesThe initial understanding of the biological effects of furocoumarins and their medical applications can be traced back to ancient times, mainly as photosensitizers for the treatment of skin disorders [15-17]. Nowadays, research on their biological and pharmacological properties has been extended to a wide range of fields, such as anticancer activities, antimicrobial effects, antioxidant properties, antiviral activities, anti-inflammatory activities, anti-Alzheimer's disease (AD) [15-21]. Despite these attractive properties, they have also been found to be associated with some adverse side effects due to their genotoxicity and cytotoxicity [21,22].
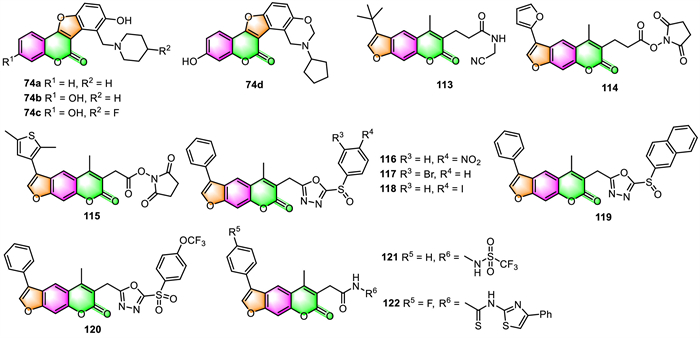
3.1. Antimicrobial activityFurocoumarins have long been known to cause multiple effects on a variety of organisms. In recent years, Yu's group commenced a series of studies [87-90] aimed at developing novel coumestan derivatives 74, furocoumarin analogs with tetracyclic skeletons, as polyketide synthase 13 (Pks13) inhibitors against mycobacterium tuberculosis (Mtb). A great number of coumestan derivatives have been synthesized by this group and evaluated for their anti-tuberculosis (anti-TB) activities against Mtb strain H37Rv, using a microplate alamar blue assay (MABA). Among those coumestan derivatives, some compounds, such as 74a–74d (Fig. 2), exhibited outstanding anti-TB against Mtb strains with MIC values of 0.125 [87], 0.0078 [88], 0.0625 [90], and 0.0039 µg/mL [90], respectively. Moreover, these four compounds are all orally bioavailable as shown in mouse models via serum inhibition titration (SIT) assay. On the basis of whole genome sequencing, the authors confirmed that coumestan derivatives could efficiently target the thioesterase domain of Mtb Pks13, making them potential Mtb Pks13 inhibitors.

|
Download:
|
| Fig. 2. Representative furocoumarins with antimicrobial activity. | |
{kind=link}
Rožman and colleagues identified 15 compounds from a library of 92 psoralen derivatives that inhibited the Mtb proteasome at low micromolar doses, with IC50 values ranging of 2–40 µmol/L [111]. In a fluorescence-based enzymatic assay, the most potent psoralens, namely 113, 114, and 115, demonstrated a mixed kind of inhibition and good overall inhibitory effectiveness with IC50 values of 3.7, 8.8, and 3.2 µmol/L, respectively. Psoralen 113 was discovered to be a reversible inhibitor with Ki (inhibition constant) values of 5.6 µmol/L (α = 0.19). Psoralens 114 and 115, on the other hand, inhibited the enzyme irreversibly with Ki values of 4.2 µmol/L (α = 6.67) and 1.1 µmol/L (α = 6.94 × 1016), respectively.
More recently, Fan's group conducted a series of investigations [32,42] on the synthesis and fungicidal evaluation of psoralen-based derivatives with various heteroatom-containing side chains. In 2022, they used a computer-aided pesticide molecular design technique to create a number of new psoralen derivatives with 1,3,4-oxadiazole moiety [42]. The fungicidal activities of the target compounds were in vitro evaluated against seven phytopathogens at a concentration of 50 µg/mL. The bioassay results revealed that compounds 116–120 demonstrated outstanding in vitro fungicidal activities against Botrytis cinerea (Bc) with EC50 values of 4.8, 6.3, 5.4, 3.3, and 3.9 µg/mL, respectively, and all outperformed the positive control YZK-C22 (EC50 = 13.4 µg/mL). Moreover, when determined at a concentration of 200 µg/mL, the in vitro inhibitory activities of compounds 117 and 118 were stronger than that of the positive control psoralen and had equal efficacy to another positive control pyrisoxazole. In addition, compound 118 (IC50 = 39.6 µmol/L) displayed almost equivalent inhibition of pyruvate kinase (PK) of Bc (BcPK) in the enzymatic assays compared to the positive control YZK-C22 (IC50 = 32.4 µmol/L), implying that compound 118 may be exploited as a novel fungicide leads with PK as the target for further structural optimization.
In a similar fashion, the same group prepared 45 psoralen derivatives with sulfonohydrazide or acyl thiourea moiety and then screened them for in vitro antifungal activities against seven phytopathogens [32]. The results showed that these prepared psoralens showed certain-to-high fungicidal efficacies. Particularly, compounds 121 and 122 had EC50 values of 12.49 µg/mL and 9.09 µg/mL against Bc, respectively, demonstrating their outstanding fungicidal activities. Further molecular docking results confirmed that these two compounds can be efficiently docked into the active site of the enzyme BcPK. In addition, some of the sulfonohydrazide or acylthiourea-containing psoralen derivatives could be leads to the development of novel fungicides with high activities.
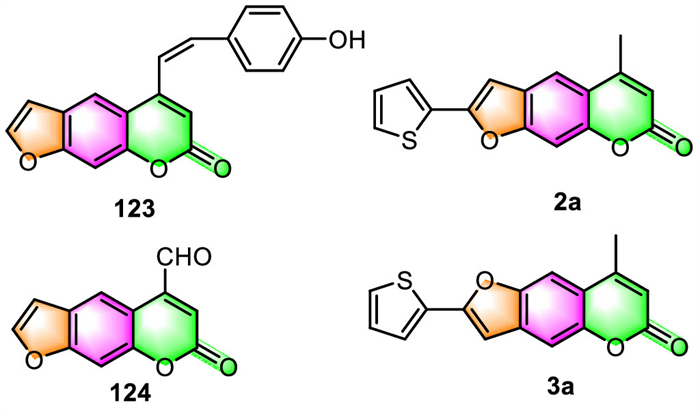
3.2. Antiviral activityXie et al. have prepared eleven 5-Schiff base substituted furocoumarin derivatives and then in vitro evaluated their promotion in melanogenesis in B16 cells and anti-bacterial properties against three species of bacteria [40]. The results showed that, when compared to 8-MOP (positive control, activation rate of 136%), compound 123 (237%) was observed to significantly enhance the amount of melanin by more than 1.7 times (Fig. 3). Also impressively, compound 124 exhibited not only stronger potency against C. albicans than the positive control (Amphotericin B), but also broad-spectrum anti-bacterial activity towards E. coli and S. aureus.

|
Download:
|
| Fig. 3. Representative furocoumarins with antiviral activity. | |
{kind=link}
In 2021, Luo's group revealed that, among all the 2-aryl furocoumarins they synthesized, 2-thiophenyl furanocoumarin 2a exerted excellent photoactivated insecticidal activity to the fourth instar larvae of Aedes aegypti (A. aegypti) [44]. The insecticidal activity tests indicated that the LC25, LC50, and LC75 concentrations of the compound in A. aegypti larvae were 53.96, 64.99, and 78.27 mg/L, respectively, following a 48-h treatment. Under UVA radiation, 2-thiophenyl furocoumarin can cause the midgut cells to produce excessive reactive oxygen species (ROS), which then block antioxidant enzymes, ultimately leading to the apoptosis of the midgut tissue and eventually to the death of the A. aegypti larva.
Interestingly, the neofurocoumarin isomer (3a) of 2a also exhibited outstanding photoactivated insecticidal activity. Similarly, under UVA irradiation, compound 3a induced a significant increase in the level of ROS in Spodoptera frugiperda (Sf9) cells, turned on the mitochondrial apoptotic signaling pathway, and finally suppressed the proliferation of Sf9 cells [45]. Thus, the same authors concluded that 2a and 3a can both be developed further as potential biochemical insecticides.
3.3. Anticancer activityNumerous studies conducted in vitro and in vivo have shown that furocoumarins exert a significant inducing apoptosis effect on a variety of cancer cell lines [16].
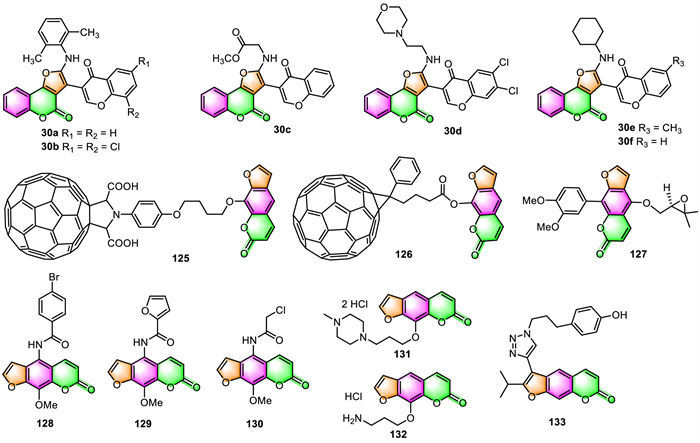
After successfully synthesizing a series of furocoumarin-chromone conjugates 30, Meydani and co-workers further evaluated their in vitro cytotoxicities against human breast cancer cells (MDA-MB-231) using the MTT assay by employing 3-formylchromone as the reference compound [63]. Most of the prepared compounds were more potent than 3-formylchromone, with IC50 values ranging from 2.56 µg/mL to 11.44 µg/mL for 30a–30f (Fig. 4) against MDA-MB-231 cells, while the 3-formylchromone reference showed IC50 values of 25.50 µg/mL. Furthermore, molecular docking studies on cyclooxygenase enzymes (COX-1, COX-2) indicated that these furocoumarin-chromone conjugates could dock into COX-1 and COX-2 successfully and formed one to three hydrogen bonds with amino acid residues in the active site of both enzymes. The two active Michael acceptor sites in the structures of these furocoumarin-chromone conjugates may contribute to their significant cytotoxicity activities.

|
Download:
|
| Fig. 4. Representative furocoumarins with anticancer activity. | |
{kind=link}
Hashimoto and co-workers have prepared two psoralen-linked fullerene derivatives (125 and 126) and evaluated their in vitro biological activities [24]. The results revealed that, under UVA irradiation, both of them possess ROS-producing activity and DNA-cleaving activity. At high concentrations and/or strong UVA irradiation, they both exhibited cytotoxic activities toward HeLa and A549 cancer cell lines. Psoralen-linked fullerenes 125 and 126 are suggested by the authors as potential photodynamic therapy (PDT) reagents.
Ohnuma and co-workers found that compound 127, a phenylfurocoumarin derivative prepared by themselves, could significantly decrease the IC50 of SN-38 in HCT-116/BCRP colon cancer cells in a dose-dependent manner, and interact with the substrate-binding site of ABCG2 (ATP-binding cassette subfamily G member 2) to decrease its ability to transport drugs [27]. The findings reveal that the furocoumarin derivative 127 may be a novel inhibitor candidate on ABCG2 and could be considered as a potential chemotherapeutic agent for overcoming ABCG2-mediated multidrug resistance (MDR) and thus improving the efficiency of cancer chemotherapy.
Khotavivattana and co-workers have synthesized twenty psoralen derivatives by modifying the 5-position of 8-MOP [30]. Using the MTT assay, these prepared compounds were then screened for their in vitro dark- and light-activated cytotoxic effects against three breast cancer cell lines: MDA-MB-231, T47-D, and SK-BR-3. Structure-activity relationship (SAR) results revealed that 4-bromobenzyl amide derivative (128) exhibited the highest dark cytotoxicity against T47-D (IC50 = 10.14 µmol/L), whereas the reference drugs doxorubicin, tamoxifen citrate, and lapatinib showed IC50 values of 1.46, 20.86, and 9.78 µmol/L, respectively. On the other hand, furanylamide (129) showed the greatest phototoxicity towards SK-BR-3 cells with the IC50 of 2.71 µmol/L, which is nearly a tenfold increase over the parent compound, methoxsalen. In addition, these furocoumarins also showed extreme selectivity for HER2+(SK-BR-3) breast cancer cell lines as opposed to HER2-(MDA-MB-231) cell lines, which was well consistent with the findings of the molecular docking investigation.
In order to enhance the cytotoxicity of methoxsalen, Guillon and co-workers synthetically incorporated seven anti-cancer pharmacophores onto the 5-position of the 8-MOP backbone [112]. The target compounds were evaluated for both dark- and light-catalyzed cytotoxicity against PAM212 keratinocyte cell line in culture. The results showed that three of the target furocoumarins with the triazeno group [–N = N–N(Me)(nBu)], the aryl azido group [–NH–CO–Ph–N3-p], and the chloroacetamido group [–NHCOCH2Cl] substituents respectively at the 5-position, displayed phototoxicity with UVA exposure, but all lower than their parent methoxsalen. Compound 130 was also found possessing dark-reaction cytotoxicity in keratinocyte culture.
In an effort to gain SAR insights and develop psoralens with enhanced potency, a library of 73 novel psoralen derivatives has been synthesized and screened by Buhimschi and co-workers using modern medicinal chemistry approaches [113]. All psoralen derivatives were evaluated for their cytotoxicities (±UVA) against B16 murine melanoma cells using the WST-1 assay. Among all derivatives tested, two psoralens (131 and 132) containing positively charged substituents turned out to be even more cytotoxic than 4′-aminomethyl-4,5′,8-trimethylpsoralen (AMT), one of the strongest psoralens ever discovered. The authors believed that these two most potent derivatives (131 and 132) may be attractive alternatives to AMT in X-PACT (X-ray Psoralen Activated Cancer Therapy). In addition, psoralen-DNA photoadduct formations of several most potent psoralens were also screened using MALDI-TOF MS. The results revealed that for many compounds, higher DNA adduct formation resulted in higher cytotoxicity.
Ivanov and co-workers prepared five 1,2,3-triazolyl-modified furocoumarins bearing fragments of hindered phenols in the side chain [47]. The cytotoxic activities of these derivatives were then examined against four human cancer cell lines, namely breast cancer (MCF-7), glioblastoma multiform cells (U-87 MG), human lung carcinoma (A-549), hepatocellular carcinoma (HepG2), using the MTT assay. The results revealed that all of them exhibited significant cytotoxic effects on the U-87 MG. Moreover, compound 133 showed nonspecific cytotoxic activity concerning all the tested cell lines, with IC50 values between 12.45 and 66.41 µmol/L.
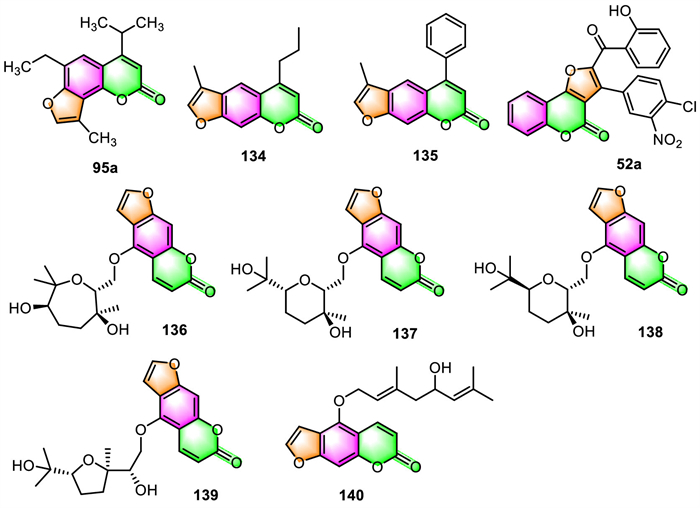
3.4. Anti-inflammatory activityIn 2018, Marzaro and co-workers reported that the isopropyl analog of trimethylangelicin (TMA) 95a prepared by them might be a superior anti-inflammatory agent to TMA (Fig. 5) [101]. Although it exhibited TMA-like inhibitory activity on NF-κB/DNA interactions with an IC50 value of 7.4 µmol/L, it does not display the side effects associated with TMA. Differently from TMA, compound 95a was found to be failed to photoreact with pyrimidine bases of DNA and therefore could not cause DNA damage. Furthermore, even at a concentration of 200 µmol/L, 95a was unable to inhibit the proliferation of IB3–1 cells. Therefore, compound 95a was considered by the authors as a potential anti-inflammatory agent without phototoxic and mutagenic side effects.

|
Download:
|
| Fig. 5. Representative furocoumarins with anti-inflammatory activity. | |
{kind=link}
Timonen and co-workers revealed that the 12 psoralen derivatives synthesized by them displayed anti-inflammatory activities via the inhibition of iNOS and IL-6 expression [43]. However, the substitution at the 4-position appeared to be necessary for their mode of action: 4-propyl psoralen 134 inhibited the NF-κB-mediated transcription, while 4-phenyl psoralen 135 suppressed iNOS mRNA expression in a posttranscriptional manner.
Fattah et al. checked the anti-inflammatory effects of the synthesized target furo[3,2-c]coumarins against LPS-induced nitric oxide (NO) production in RAW 264.7 macrophages using the Griess assay [77]. Results showed that their anti-inflammatory properties were incomparably superior to the efficacy of the positive control indomethacin (IC50 value of 212 ± 8 µmol/L). Impressively, compound 52a was identified as possessing the best inhibitory activity with the minimal IC50 value of 4.2 ± 0.3 µmol/L.
A group of feroniellins, which contain a furocoumarin skeleton coupled to monoterpenic five- to seven-membered ethereal rings by an ether linker, have been synthesized by Nishikawa et al. They started from easily available bergamottin using a “ring size-divergent” synthetic strategy [29]. These synthesized target compounds 136–139 (Fig. 5) were proved to display NO production inhibitory activities in LPS-stimulated RAW264 cells (approximately 20%–60% at 100 µmol/L). None of them was found to be cytotoxic even up to a concentration of 100 µmol/L.
Huang and co-workers revealed that notopterol 140, a natural linear furocoumarin, can significantly reduce the expression of IL-1β and IL-6 (the pro-inflammatory factors) and proliferating cell nuclear antigen (PCNA) in the lungs of pulmonary arterial hypertension (PAH) rats [114]. The anti-inflammatory and anti-proliferative properties of 133 allow it to be used therapeutically in the treatment of PAH.
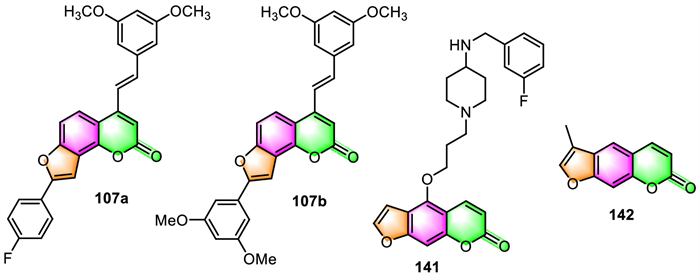
3.5. Anti-Alzheimer's disease and anti-Parkinsonian reagentAgbo and co-workers have reported that within all the furocoumarin-stilbene hybrids 107 they prepared, 107a displayed the most anticholinesterase activity and β-secretase inhibitory effect, with IC50 values of 1.8, 3.5, and 9.9 µmol/L against acetylcholinesterase (AChE), butyrylcholinesterase (BChE), and β-secretase, respectively (Fig. 6) [108]. While 107b was found to be the most active towards cyclooxygenase-2 (COX-2) and lipoxygenase-5 (LOX-5), with IC50 values of 8.6 and 13.9 µmol/L, respectively. Therefore, compounds 107a and 107b have been identified as potential multifunctional drugs against multiple biochemical targets closely linked to AD.

|
Download:
|
| Fig. 6. Representative furocoumarins for treatment of AD or PD. | |
{kind=link}
Jiang's team published research on the synthesis and biological assessment of the notopterol derivatives for the treatment of AD [31]. Among the 48 notopterol derivatives prepared, 141 possessed triple inhibitory activity against AChE, BACE1, and GSK3β in enzymatic assays, with IC50 values of 1 ± 0.4, 20 ± 1, and 15 ± 1 µmol/L, respectively. Additionally, 141 demonstrated acceptable bioavailability, good oral safety profile (F = 9.8%), and adequate blood-brain barrier (BBB) penetrability. Impressively, oral administration of 141 to Aβ-induced AD mice can significantly improve its memory and learning deficits, and the expression levels of Aβ-related proteins were significantly decreased in the AD mice after 141 treatment.
Parkinson's disease (PD) is ranked as the second most frequent neurodegenerative disease (ND) following AD. In 2019, Olaya and co-workers evaluated the antiparkinsonian activity, inhibitory activity towards monoamine oxidases (MAO), and antioxidant activity of 142 [115]. The results showed that hypokinesia was significantly reversed in the reserpine and levodopa models at a dosage of 100 mg/kg of 142. This effect could be attributed to 142′s selective inhibitory activity against the human monoamine oxidase hMAO-B isoform (IC50 value of 41.63 ± 2.79 µmol/L).
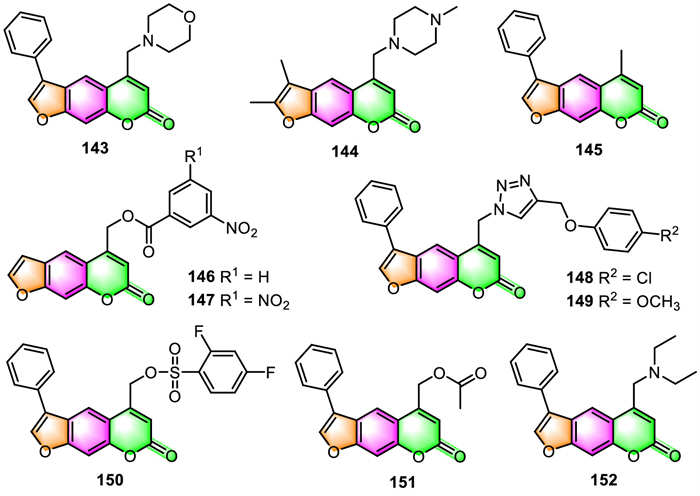
3.6. Skin protection and treatmentAisa and colleagues have recently carried out a variety of related studies [35,36,39,41,116-118] to explore bioactive furocoumarin derivatives with potential anti-vitiligo properties. They have successively prepared several classes of furocoumarins via total synthesis or structural modification, and their melanin synthesis and tyrosinase activities were evaluated in murine B16 cells, and SAR was also investigated. Results indicated that many furocoumarin derivatives displayed better activities toward melanin synthesis than the positive control (8-MOP) in B16 melanoma cells in a concentration-dependent manner. Among them, compounds 143–152 (Fig. 7) were respectively identified as the most promising candidate derivatives for the further pharmacological study of anti-vitiligo [35,36,39,41,116-118]. Moreover, the different molecular functions in melanogenesis investigations revealed that these compounds stimulate melanin biosynthesis through various signaling pathways: 145 proceeds through activation of p38 mitogen-activated protein kinase (p38 MAPK) and the protein kinase A (PKA) signaling pathways [116], and 146 [36] and 143 [117] by up-regulating MITF and TYR family via Akt/GSK3β/β-catenin signaling pathways, 152 [118] turns on the activation of cAMP/PKA and MAPKs signal pathway, while compounds 150 [41] and 151 [41] stimulates the p38 MAPK and Akt/GSK3β/β-catenin signaling routes.

|
Download:
|
| Fig. 7. Representative furocoumarins for skin protection and treatment. | |
{kind=link}
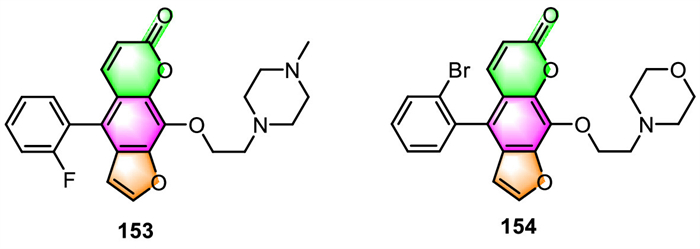
He's group revealed that almost all of the 5-phenyl furocoumarin derivatives they synthesized have been found to cause moderate to significant vasodilatation in the isolated rat mesenteric artery (MA), basilar artery (BA), and renal artery (RA) in vitro [23]. Compound 153 exhibited the highest vasodilatory activity in MA, with the EC50 value of 0.56 µmol/L (Fig. 8). The molecular docking analysis revealed that the combination of 153 and proteins can be affected by both the bulky side chain and the fluorine substituent.

|
Download:
|
| Fig. 8. Representative furocoumarins with vasodilatory activity. | |
{kind=link}
The same group recently demonstrated that these kinds of 5-phenyl furocoumarin derivatives could also alleviate allergic reactions and disorders [119]. They drastically inhibited MRGPRX2 (Mas-related G protein-coupled receptor X2) agonist-induced degranulation and cytokine release in LAD2 cells, while also relieving local and systemic anaphylaxis in mice. The IC50 value of 154 (IC50 = 12.3 µmol/L), the most effective derivative, is less than one-tenth that of the parent compound imperatorin (IC50 = 159.80 µmol/L). Further research into possible targets indicated that 154 has anti-pseudo-allergic action through both the MRGPRX2 and IgE receptors. These findings imply that 154 could be utilized to treat mast cell-dependent allergy diseases.
3.8. OthersIn addition to the biomedical applications mentioned above, furocoumarin derivatives also exhibit other excellent properties, particularly photophysical properties. The nucleus of furocoumarins is constructed by an electron-rich furan ring and an electron-deficient pyrone ring conjugated with a benzene ring. This structural characteristic provides furocoumarins with the intrinsic donor-acceptor chromophore and tunable photophysical properties [26,28,64-66,73,81,120-121], which allows them to be used as fluorescent probes for the detection of analytes or efficient photosensitizers for bio-medical applications [122,123].
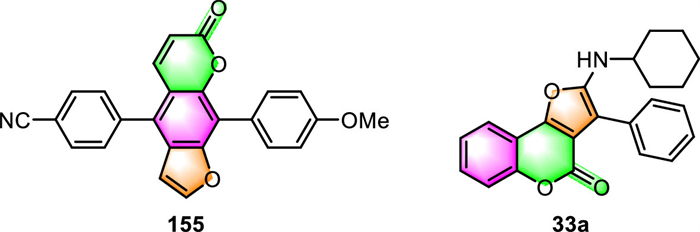
For example, Geenen et al. have synthesized novel donor-acceptor psoralen cruciforms containing an interesting X-shaped chromophore and investigated their photophysical characteristics [26]. This psoralen derivative 155 (Fig. 9) demonstrated high fluorescence quantum yields of up to 38% and large Stokes shifts of up to 8900 cm−1. Moreover, some of them exhibited remarkable solvatochromic, acidochromic, and aggregation-induced emission (AIE) properties.

|
Download:
|
| Fig. 9. Representative furocoumarins with photophysical properties. | |
{kind=link}
Sarih and co-workers developed a novel fluorescent ratiometric chemosensor 33a based on furo[3,2-c]coumarin [65]. This fluorescence sensor has a high fluorescence quantum yield (ФF = 0.48), a long fluorescence lifetime (5.6 ns), and a large Stokes shift. Even in the presence of other metal ions, 33a demonstrates a highly selective, sensitive, and immediate turn-off fluorescence response to Fe3+. The detection limit of this sensor was measured to be as low as 1.93 µmol/L, which was quite competitive with many other reported Fe3+ sensors.
4. Conclusions and outlookDuring the last 5 years, furocoumarin has remained one of the most concerned heterocyclic compounds, attracting the interest of many synthetic chemists and pharmaceutical chemists. Numerous novel synthetic methods and technologies as well as versatile starting materials have emerged to construct diverse furocoumarin derivatives. However, research on the synthesis of each class of the furocoumarin family is unbalanced; among the 16 members, a large number of furo[3,2-g]coumarins, furo[3,2-c]coumarins, furo[2,3-h]coumarins, and furo[3,4-c]coumarins derivatives have been synthesized, while other members were rarely reported. In parallel with the development of the synthetic methodology, research in cell cultures and animal models has been conducted to comprehend the physicochemical features of these newly synthesized furocoumarins. They have been proven to have anti-cancer, antimicrobial, antiviral, anti-inflammatory, anti-AD, anti-PD, and vasodilatory properties, respectively, and some of the most active compounds have the potential for further clinical applications.
Despite the significant progress made, there remain many opportunities for further exploration of furocoumarin derivatives. Firstly, the synthesis and biological activity of those long-neglected members of the furocoumarin family, such as furo[2,3-c]coumarins, furo[3,4-g]coumarins, furo[3,2-h]coumarins, furo[3,4-h]coumarins, and furo[f]coumarins is highly desirable. Furthermore, from the perspective of green chemistry and pharmaceutical applications, furocoumarin synthesis using recently developed technologies such as organic electrosynthesis [124], visible-light-induced photoredox catalysis [125], chemoenzymatic synthesis, and continuous-flow synthesis [126] may be a promising direction for future research. Additionally, more detailed action mechanistic studies are required to have a thorough knowledge of the molecular foundation for the biological roles of furocoumarin derivatives in living systems. Finally, the genotoxic effects of potent furocoumarin derivatives must be carefully considered in order to obtain a balanced optimization of pharmacological effects toward multiple targets. It is highly anticipated that more and more furocoumarins with outstanding performance will be developed and play an increasingly essential role in pharmaceutical chemistry in the coming years.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is dedicated to Professor Jiannan Xiang on the occasion of his 65th birthday. We are grateful for financial support from the National Natural Science Foundation of China (Nos. 21971211 and 22171232), the Natural Science Foundation of Zhejiang Province (No. 2022XHSJJ007), the Qiantang River Talent Foundation (No. QJD1902029), and Westlake University. This research was supported by Instrumentation and Service Centers for Molecular Science and for Physical Science (ISCMS and ISCPS), respectively, as well as by the Mass Spectrometry & Metabolomics Core Facility at the Center for Biomedical Research Core Facilities and Westlake University HPC Center.
| [1] |
M. Lončarić, D. Gašo-Sokač, S. Jokić, et al., Biomolecules 10 (2020) 151. DOI:10.3390/biom10010151 |
| [2] |
F. Annunziata, C. Pinna, S. Dallavalle, et al., Int. J. Mol. Sci. 21 (2020) 4618. DOI:10.3390/ijms21134618 |
| [3] |
S. Ponra, K.C. Majumdar, RSC Adv. 6 (2016) 37784-37922. DOI:10.1039/C5RA27069C |
| [4] |
A. Deepthi, B.P. Babu, A.L. Balachandran, et al., Org. Prep. Proced. Int. 51 (2019) 409-442. DOI:10.1080/00304948.2019.1633228 |
| [5] |
V. Cadierno, Metal-catalyzed routes for the synthesis of furocoumarins and coumestans, in: G. Brahmachari (Ed.), Green Synthetic Approaches For Biologically Relevant Heterocycles, Elsevier, Amsterdam, 2015, pp. 77–100.
|
| [6] |
V. Cadierno, Metal-catalyzed routes for the synthesis of furocoumarins and coumestans, in: G. Brahmachari (Ed.), Green Synthetic Approaches For Biologically Relevant Heterocycles, 2nd Ed., Elsevier, Amsterdam, 2021, pp. 53–96.
|
| [7] |
V.F. Traven, Molecules 9 (2004) 50-66. DOI:10.3390/90300050 |
| [8] |
L. Santana, E. Uriarte, F. Roleira, et al., Curr. Med. Chem. 11 (2004) 3239-3261. DOI:10.2174/0929867043363721 |
| [9] |
N. Singh, K. Rajotiya, N. Lamba, et al., Curr. Org. Chem. 26 (2022) 324-341. DOI:10.2174/1385272826666220126155703 |
| [10] |
E.R. El-Sawy, A.B. Abdelwahab, G. Kirsch, et al., Molecules 26 (2021) 483. DOI:10.3390/molecules26020483 |
| [11] |
I. Cortés, L.J. Cala, A.B.J. Bracca, et al., RSC Adv. 10 (2020) 33344-33377. DOI:10.1039/d0ra06930b |
| [12] |
F.G. Medina, J.G. Marrero, M. Macías-Alonso, et al., Nat. Prod. Rep. 32 (2015) 1472-1507. DOI:10.1039/C4NP00162A |
| [13] |
S. Pasricha, K. Mittal, P. Gahlot, et al., J. Iran. Chem. Soc. 19 (2022) 4035-4092. DOI:10.1007/s13738-022-02603-x |
| [14] |
G. Patel, S. Banerjee, Curr. Org. Chem. 24 (2020) 2566-2587. DOI:10.2174/1385272824999200709125717 |
| [15] |
R. Gambari, I. Lampronti, N. Bianchi, et al., Structure and biological activity of furocoumarins, in: M.T.H. Khan (Ed.), Bioactive Heterocycles Ⅲ. Topics in Heterocyclic Chemistry (vol. 9), Springer, Berlin, Heidelberg, 2007, pp. 265–276.
|
| [16] |
S. Ahmed, H. Khan, M. Aschner, et al., Int. J. Mol. Sci. 21 (2020) 5622. DOI:10.3390/ijms21165622 |
| [17] |
R. Bruni, D. Barreca, M. Protti, et al., Molecules 24 (2019) 2163. DOI:10.3390/molecules24112163 |
| [18] |
Y. Ren, X. Song, L. Tan, et al., Front. Pharmacol. 11 (2020) 571535. DOI:10.3389/fphar.2020.571535 |
| [19] |
A. Wu, J. Lu, G. Zhong, et al., Phytother. Res. 36 (2022) 3805-3832. DOI:10.1002/ptr.7577 |
| [20] |
C.K. Mahendra, L.T.H. Tan, W.L. Lee, et al., Front. Pharmacol. 11 (2020) 366. DOI:10.3389/fphar.2020.00366 |
| [21] |
M.M. Quetglas-Llabrés, C. Quispe, J. Herrera-Bravo, et al., Oxid. Med. Cell. Longev. 2022 (2022) 8615242. |
| [22] |
M. Deng, L. Xie, L. Zhong, et al., Eur. J. Pharmacol. 879 (2020) 173124. DOI:10.1016/j.ejphar.2020.173124 |
| [23] |
D. Wei, Y.J. Hou, Y.T. Xie, et al., J. Asian Nat. Prod. Res. 22 (2020) 153-166. DOI:10.1080/10286020.2018.1540600 |
| [24] |
A. Hashimoto, T. Takamura-Enya, Y. Oda, Photochem. Photobiol. 95 (2019) 1403-1411. DOI:10.1111/php.13138 |
| [25] |
B.Z. Zsidó, M. Balog, N. Erős, et al., Int. J. Mol. Sci. 21 (2020) 508. DOI:10.3390/ijms21020508 |
| [26] |
S.R. Geenen, T. Schumann, T.J.J. Müller, J. Org. Chem. 85 (2020) 9737-9750. DOI:10.1021/acs.joc.0c01059 |
| [27] |
S. Kokubo, S. Ohnuma, M. Murakami, et al., Int. J. Mol. Sci. 22 (2021) 12502. DOI:10.3390/ijms222212502 |
| [28] |
J. Bertling, K.A. Thom, S. Geenen, et al., Photochem. Photobiol. 97 (2021) 1534-1547. DOI:10.1111/php.13480 |
| [29] |
K. Nishikawa, T. Niwa, K. Nishikibe, et al., Chem. Eur. J. 27 (2021) 11045-11049. DOI:10.1002/chem.202101603 |
| [30] |
C. Aekrungrueangkit, S. Wangngae, A. Kamkaew, et al., Sci. Rep. 12 (2022) 13487. DOI:10.1038/s41598-022-17625-x |
| [31] |
N. Wang, W. Liu, L. Zhou, et al., ACS Omega 7 (2022) 32131-32152. DOI:10.1021/acsomega.2c03368 |
| [32] |
J. Dong, K. Li, Z. Hong, et al., Mol. Divers. 7 (2023) 571-588. DOI:10.1007/s11030-022-10402-y |
| [33] |
Z. Qin, M. Zhao, K. Zhang, et al., J. Org. Chem. 86 (2021) 7864-7871. DOI:10.1021/acs.joc.1c00776 |
| [34] |
M. Tao, A. Wang, P. Guo, et al., Adv. Synth. Catal. 364 (2022) 24-29. DOI:10.1002/adsc.202100940 |
| [35] |
C. Niu, D. Zang, H.A. Aisa, Chem. Res. Chin. Univ. 34 (2018) 408-414. DOI:10.1007/s40242-018-7338-4 |
| [36] |
C. Niu, L. Yin, H.A. Aisa, Int. J. Mol. Sci. 19 (2018) 746. DOI:10.3390/ijms19030746 |
| [37] |
E.S. Schiffrer, I. Sosic, A. Sterman, et al., MedChemComm 10 (2019) 1958-1965. DOI:10.1039/c9md00365g |
| [38] |
C.Y. Chen, T.H. Yang, C.D. Pan, et al., J. Carbohyd. Chem. 38 (2019) 179-191. DOI:10.1080/07328303.2019.1609018 |
| [39] |
C. Niu, X. Lu, H.A. Aisa, RSC Adv. 9 (2019) 1671-1678. DOI:10.1039/c8ra09755k |
| [40] |
H. Xie, C. Niu, Z. Chao, et al., Heterocycl. Commun. 26 (2020) 176-184. DOI:10.1515/hc-2020-0115 |
| [41] |
C. Niu, D. Zang, H.A. Aisa, Int. J. Mol. Sci. 23 (2022) 7959. DOI:10.3390/ijms23147959 |
| [42] |
J. Dong, W. Gao, K. Li, et al., J. Agric. Food Chem. 70 (2022) 3435-3446. DOI:10.1021/acs.jafc.1c07911 |
| [43] |
J.M. Timonen, K. Vuolteenaho, T. Leppänen, et al., J. Heterocyclic Chem. 55 (2018) 2590-2597. DOI:10.1002/jhet.3318 |
| [44] |
J. Wu, L. Wang, Y. Zhang, et al., J. Agric. Food Chem. 69 (2021) 1091-1106. DOI:10.1021/acs.jafc.0c07237 |
| [45] |
X. Shao, Z. Zhang, X. Qian, et al., Toxins 14 (2022) 677. DOI:10.3390/toxins14100677 |
| [46] |
S.A. Kremis, D.S. Baev, A.V. Lipeeva, et al., J. Biochem. Mol. Toxicol. 33 (2019) e22396. |
| [47] |
A.A. Ivanov, E.A. Ukladov, S.A. Kremis, et al., Protoplasma 259 (2022) 1321-1330. DOI:10.1007/s00709-022-01739-0 |
| [48] |
D.I. Brahmbhatt, J.M. Gajera, C.N. Patel, et al., J. Heterocycl. Chem. 43 (2006) 1699-1702. DOI:10.1002/jhet.5570430643 |
| [49] |
B. Borah, K.D. Dwivedi, R. Chowhan, Asian J. Org. Chem. 10 (2021) 3101-3126. DOI:10.1002/ajoc.202100550 |
| [50] |
G.M. Mohammadi Ziarani, R. Moradi, T. Ahmadi, et al., Mol. Divers. 23 (2019) 1029-1064. DOI:10.1007/s11030-019-09918-7 |
| [51] |
X. Chu, Z. Tang, J. Ma, et al., Tetrahedron 74 (2018) 970-974. DOI:10.1016/j.tet.2018.01.012 |
| [52] |
S. Borthakur, P.P. Kaishap, S. Gogoi, Asian J. Org. Chem. 7 (2018) 918-921. DOI:10.1002/ajoc.201800161 |
| [53] |
J. Kim, K. Lee, P.H. Lee, Bull. Korean Chem. Soc. 41 (2020) 709-718. DOI:10.1002/bkcs.12058 |
| [54] |
T.A. To, Y.H. Vo, A.T. Nguyen, et al., Org. Biomol. Chem. 16 (2018) 5086-5089. DOI:10.1039/C8OB01064A |
| [55] |
M. He, Z. Yan, W. Wang, et al., Tetrahedron Lett. 59 (2018) 3706-3712. DOI:10.1016/j.tetlet.2018.09.007 |
| [56] |
Q.T. Pham, P.Q. Le, H.V. Dang, et al., RSC Adv. 10 (2020) 44332-44338. DOI:10.1039/d0ra07566c |
| [57] |
S.A.Z. Ahmad, F.A. K.han, Synlett 34 (2023) 823-828. DOI:10.1055/a-1912-3884 |
| [58] |
V.V. Pelipko, R.I. Baichurin, K.A. Lyssenko, et al., Mendeleev Commun. 32 (2022) 454-456. DOI:10.1016/j.mencom.2022.07.009 |
| [59] |
P.H. Pham, Q.T.D. Nguyen, N.K.Q. Tran, et al., Eur. J. Org. Chem. 32 (2018) 4431-4435. DOI:10.1002/ejoc.201800983 |
| [60] |
M. Patel, P. Parikh, J. Timaniya, et al., Arkivoc 5 (2020) 155-167. DOI:10.24820/ark.5550190.p011.170 |
| [61] |
S. Rani, N. Kamra, S. Thakral, et al., J. Heterocycl. Chem. 59 (2022) 144-160. DOI:10.1002/jhet.4374 |
| [62] |
V. Nair, R.S. Menon, A.U. Vinod, et al., Tetrahedron Lett. 43 (2002) 2293-2295. DOI:10.1016/S0040-4039(02)00226-5 |
| [63] |
A. Meydani, S. Yousefi, R. Gharibi, et al., ChemistrySelect 4 (2019) 3315-3324. DOI:10.1002/slct.201900009 |
| [64] |
N. Muhamad Sarih, P. Myers, A. Slater, Sci. Rep. 9 (2019) 11834. DOI:10.1038/s41598-019-47847-5 |
| [65] |
N.M. Sarih, A. Ciupa, S. Moss, et al., Sci. Rep. 10 (2020) 7421. DOI:10.1038/s41598-020-63262-7 |
| [66] |
N.N.M.Y. Chan, A. Idris, Z.H.Z. Abidin, Mater. Chem. Phys. 276 (2022) 125406. DOI:10.1016/j.matchemphys.2021.125406 |
| [67] |
N. Kerru, L. Gummidi, K.K. G.angu, et al., ChemistrySelect 5 (2020) 4104-4110. DOI:10.1002/slct.202000796 |
| [68] |
H. Zhou, Y.W. Sun, J.B. Xu, et al., Res. Chem. Intermed. 48 (2022) 1763-1772. DOI:10.1007/s11164-022-04664-2 |
| [69] |
Y.O. Gorbunov, V.S. Mityanov, V.G. Melekhina, et al., Russ. Chem. Bull. 67 (2018) 304-307. DOI:10.1007/s11172-018-2074-y |
| [70] |
X. Chang, X. Zhang, Z. Chen, Org. Biomol. Chem. 16 (2018) 4279-4287. DOI:10.1039/c8ob00942b |
| [71] |
X. Chang, P. Zeng, Z. Chen, Eur. J. Org. Chem. 38 (2019) 6478-6485. DOI:10.1002/ejoc.201900987 |
| [72] |
Z. Chen, P. Zeng, S. Zhang, et al., ChemistrySelect 6 (2021) 4539-4543. DOI:10.1002/slct.202101029 |
| [73] |
P. Rajesh, A.I. Almansour, N. Arumugam, et al., Org. Biomol. Chem. 19 (2021) 1060-1065. DOI:10.1039/D0OB02276D |
| [74] |
A. Jana, D. Ali, P. Bhaumick, et al., J. Org. Chem. 87 (2022) 7763-7777. DOI:10.1021/acs.joc.2c00353 |
| [75] |
A.N. Komogortsev, V.G. Melekhina, B.V. Lichitsky, et al., Tetrahedron 111 (2022) 132716. DOI:10.1016/j.tet.2022.132716 |
| [76] |
S. Kolita, P. Borah, P.S. N.aidu, et al., Tetrahedron 72 (2016) 532-538. DOI:10.1016/j.tet.2015.12.016 |
| [77] |
T.A. Fattah, A. Saeed, Y.M. Al-Hiari, et al., J. Mol. Struct. 1179 (2019) 390-400. DOI:10.1016/j.molstruc.2018.11.014 |
| [78] |
W.E. Noland, H.V. Kumar, A. Sharma, et al., Org. Lett. 22 (2020) 1801-1806. DOI:10.1021/acs.orglett.0c00123 |
| [79] |
H. Abbasi-Dehnavi, M. Ghashang, Heterocycl. Commun. 24 (2018) 19-22. DOI:10.1515/hc-2017-0141 |
| [80] |
D. Wagare, M. Shaikh, D. Lingampalle, et al., Curr. Organocatalysis 8 (2021) 217-222. DOI:10.2174/2213337207999200706001142 |
| [81] |
V.N. Babu, A. Murugan, N. Katta, et al., J. Org. Chem. 84 (2019) 6631-6641. DOI:10.1021/acs.joc.9b00096 |
| [82] |
V.P. Perevalov, V.S. Mityanov, B.V. Lichitsky, et al., Tetrahedron 76 (2020) 130947. DOI:10.1016/j.tet.2020.130947 |
| [83] |
S.M. Yang, C.Y. Wang, C.K. Lin, et al., Angew. Chem. Int. Ed. 57 (2018) 1668-1672. DOI:10.1002/anie.201711524 |
| [84] |
N. Panda, I. Mattan, RSC Adv. 8 (2018) 7716-7725. DOI:10.1039/c7ra12419h |
| [85] |
L. Fu, S. Li, Z. Cai, et al., Nat. Catal. 1 (2018) 469-478. DOI:10.1038/s41929-018-0080-y |
| [86] |
S.S. Vagh, B.J. Hou, A. Edukondalu, et al., Org. Lett. 23 (2021) 842-846. DOI:10.1021/acs.orglett.0c04082 |
| [87] |
W. Zhang, S.C. Lun, S.H. Wang, et al., J. Med. Chem. 61 (2018) 791-803. DOI:10.1021/acs.jmedchem.7b01319 |
| [88] |
W. Zhang, S.C. Lun, L.L. Liu, et al., J. Med. Chem. 62 (2019) 3575-3589. DOI:10.1021/acs.jmedchem.9b00010 |
| [89] |
W. Zhang, L.L. Liu, S.C. Lun, et al., Eur. J. Med. Chem. 213 (2021) 113202. DOI:10.1016/j.ejmech.2021.113202 |
| [90] |
W. Zhang, S.C. Lun, S.S. Wang, et al., J. Med. Chem. 65 (2022) 13240-13252. DOI:10.1021/acs.jmedchem.2c01064 |
| [91] |
H. Zhang, C. Ma, Z. Zheng, et al., Chem. Commun. 54 (2018) 4935-4938. DOI:10.1039/c8cc02474j |
| [92] |
X. Song, X. Luo, J. Sheng, et al., RSC Adv. 9 (2019) 17391-17398. DOI:10.1039/c9ra01909j |
| [93] |
V.N. Shinde, K. Rangan, D. Kumar, et al., J. Org. Chem. 86 (2021) 9755-9770. DOI:10.1021/acs.joc.1c01097 |
| [94] |
Q.F. Yan, Y. Jiang, X.H. Song, et al., J. Org. Chem. 87 (2022) 5785-5794. DOI:10.1021/acs.joc.2c00120 |
| [95] |
X.H. Ouyang, F.L. Tan, R.J. Song, et al., Org. Lett. 20 (2018) 6765-6768. DOI:10.1021/acs.orglett.8b02883 |
| [96] |
Q. Li, X. He, J. Tao, et al., Adv. Synth. Catal. 361 (2019) 1874-1886. DOI:10.1002/adsc.201900012 |
| [97] |
Y.J. Wang, T.T. Wang, L. Yao, et al., J. Org. Chem. 85 (2020) 9514-9524. DOI:10.1021/acs.joc.0c00249 |
| [98] |
M.K. Pandya, M.R. Chhasatia, N.D. Vala, et al., J. Drug Deliv. Ther. 9 (2019) 32-42. |
| [99] |
Y. Dong, J.T. Yu, S. Sun, et al., Chem. Commun. 56 (2020) 6688-6691. DOI:10.1039/d0cc00176g |
| [100] |
M.L.N. Rao, S. Nand, V.N. Murty, Asian J. Org. Chem. 11 (2022) e202100604. DOI:10.1002/ajoc.202100604 |
| [101] |
G. Marzaro, I. Lampronti, E. D'Aversa, Eur. J. Med. Chem. 151 (2018) 285-293. DOI:10.1016/j.ejmech.2018.03.080 |
| [102] |
A. Carbone, A. Montalbano, V. Spanò, et al., Eur. J. Med. Chem. 180 (2019) 283-290. DOI:10.1016/j.ejmech.2019.07.025 |
| [103] |
T.V. Shokol, V.S. Moskvina, E.K. Glebov, et al., Chem. Nat. Compd. 55 (2019) 716-718. DOI:10.1007/s10600-019-02787-4 |
| [104] |
T.V. Shokol, V.S. Moskvina, Ye.K. Hlibov, et al., Chem. Nat. Compd. 57 (2021) 33-37. DOI:10.1007/s10600-021-03275-4 |
| [105] |
T. Shokol, A. Suprun, V. Moskvina, et al., French-Ukrainian J. Chem. 9 (2021) 83-93. DOI:10.17721/fujcv9i2p83-93 |
| [106] |
C. Schultze, B. Schmidt, Beilstein J. Org. Chem. 14 (2018) 2991-2998. DOI:10.3762/bjoc.14.278 |
| [107] |
T.O. Olomola, M.J. Mphahlele, J. Fluorine Chem. 229 (2020) 109395. DOI:10.1016/j.jfluchem.2019.109395 |
| [108] |
E.N. Agbo, S. Gildenhuys, Y.S. Choong, et al., Bioorg. Chem. 101 (2020) 103997. DOI:10.1016/j.bioorg.2020.103997 |
| [109] |
B.V. Lichitsky, A.N. Komogortsev, V.G. Melekhina, Molbank 2021 (2021) M1304. DOI:10.3390/m1304 |
| [110] |
R. Singh, T. Horsten, R. Prakash, et al., Beilstein J. Org. Chem. 17 (2021) 977-982. DOI:10.3762/bjoc.17.79 |
| [111] |
K. Rožman, E.M. Alexander, E. Ogorevc, et al., Molecules 25 (2020) 1305. DOI:10.3390/molecules25061305 |
| [112] |
C.D. Guillon, Y.H. Jan, N. Foster, et al., Bioorg. Med. Chem. Lett. 29 (2019) 619-622. DOI:10.1016/j.bmcl.2018.12.048 |
| [113] |
A.D. Buhimschi, D.M. Gooden, H. Jing, et al., Photochem. Photobiol. 96 (2020) 1014-1031. DOI:10.1111/php.13263 |
| [114] |
L. Huang, H. Li, S. Huang, et al., Front. Cardiovasc. Med. 9 (2022) 859422. DOI:10.3389/fcvm.2022.859422 |
| [115] |
M.P. Olaya, N.E. Vergel, J.L. López, et al., Biomédica 39 (2019) 491-501. DOI:10.7705/biomedica.4299 |
| [116] |
L. Yin, G. Pang, C. Niu, et al., Int. J. Mol. Med. 41 (2018) 3727-3735. |
| [117] |
D. Zang, C. Niu, H.A. Aisa, Drug Des. Devel. Ther. 13 (2019) 623-632. DOI:10.2147/dddt.s180960 |
| [118] |
D. Zang, C. Niu, X. Lu, et al., Int. J. Mol. Sci. 23 (2022) 14190. DOI:10.3390/ijms232214190 |
| [119] |
C. Wang, Y. Hou, S. Ge, et al., Biomed. Pharmacother. 150 (2022) 112982. DOI:10.1016/j.biopha.2022.112982 |
| [120] |
I.O. Akchurin, A.I. Yakhutina, A.Y. Bochkov, Heterocycl. Commun. 24 (2018) 85-91. DOI:10.1515/hc-2017-0253 |
| [121] |
O.N. Tchaikovskaya, N.G. Dmitrieva, E.N. Bocharnikova, et al., Front. Chem. 9 (2021) 754950. DOI:10.3389/fchem.2021.754950 |
| [122] |
M. Tang, Y. Liang, J. Liu, et al., CCS Chem. 4 (2022) 3230-3237. DOI:10.31635/ccschem.022.202101679 |
| [123] |
M. Tang, Y. Liang, J. Liu, et al., Mater. Today Chem. 24 (2022) 100868. DOI:10.1016/j.mtchem.2022.100868 |
| [124] |
L. Ma, X. Gao, X. Liu, et al., Chin. Chem. Lett. 34 (2023) 107735. DOI:10.1016/j.cclet.2022.08.015 |
| [125] |
B. Wang, L. Zou, L. Wang, et al., Chin. Chem. Lett. 32 (2021) 1229-1232. DOI:10.1016/j.cclet.2020.08.013 |
| [126] |
J. Xie, D. Zhao, Chin. Chem. Lett. 31 (2020) 2395-2400. DOI:10.1016/j.cclet.2020.03.022 |