 2023, Vol. 34
2023, Vol. 34 


b School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China;
c School of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
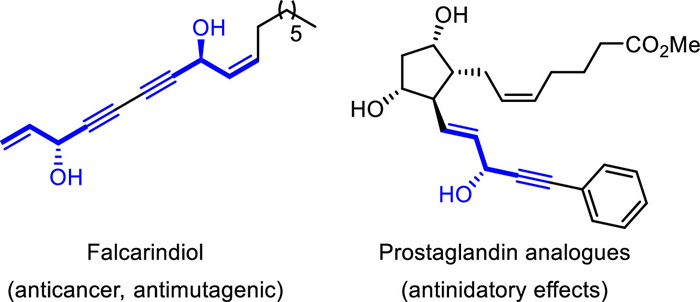
1,4-Enynes bearing internal stereogenic centers are privileged motifs in biological active molecules (Fig. 1) [1,2] and also serve as important synthetic intermediates [3-7]. In especial, due to the reliable and controllable downstream transformations of alkene and alkyne units, enantioenriched 1,4-enynes have proved to be valuable building blocks for organic synthesis. For example, with the special methylene-interrupted unit, 1,4-enynes could undergo various transformations such as stereospecific proton migration [8,9], cycloaddition [10-12], cycloisomerization [13-15], and others [16-20]. As a result, a variety of efficient synthetic methods to achieve this manifold have been developed. Despite of these great progress during the past decade in this field, related review for this emerging area on catalytic asymmetric construction of 1,4-enynes remains absent.

|
Download:
|
| Fig. 1. Bioactive molecules containing 1,4-enynes. | |
{kind=link}
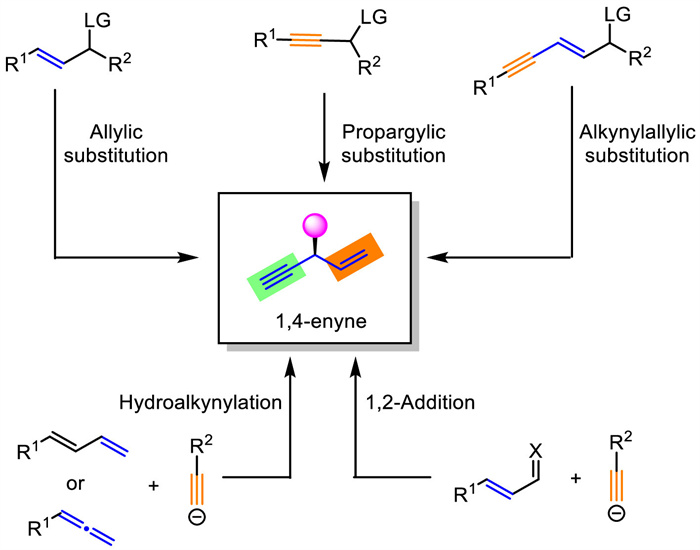
Herein, we review the reported methods and strategies towards the preparations of enantioenriched 1,4-enynes and outline related mechanisms. The synthetic methodologies are mainly categorized into five types based on the reaction design, including allylic substitutions with alkynyl nucleophiles, propargylic substitutions with alkenyl nucleophiles, alkynylallylic substitution, hydroalkynylation, and 1,2-addition of alkynes to conjugated imines/aldehydes (Scheme 1). Each section might further be divided into several subclasses based on catalyst types including transition metal catalysis and organic catalysis.

|
Download:
|
| Scheme 1. Strategies for asymmetric syntheses of 1,4-enynes. | |
{kind=link}
2. Asymmetric allylic substitution with alkynyl nucleophiles 2.1. Cu catalysis
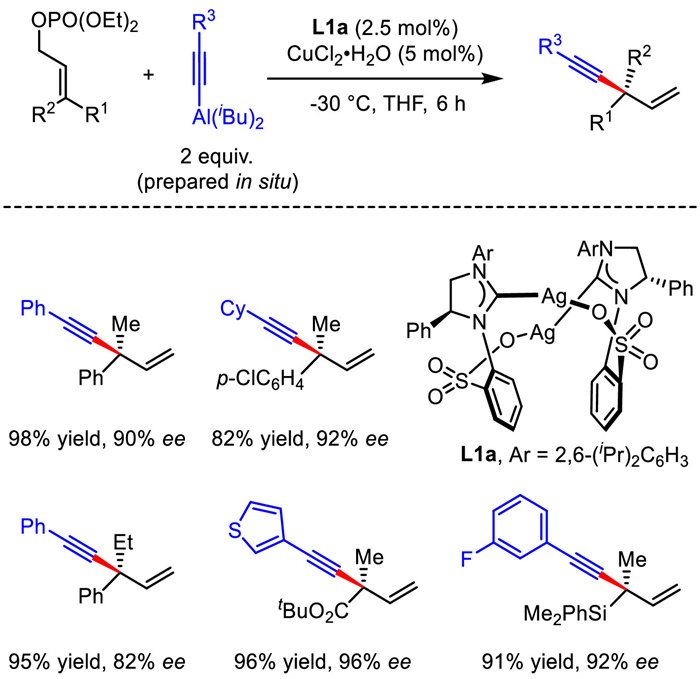
Asymmetric allylic substitution reactions are among the most important methods for constructing a series of stereogenic centers [21-25]. Undoubtedly, asymmetric allylic substitution turns to be a prevalent strategy for the synthesis of 1,4-enynes. Although Kobayashi developed copper-involved SN2-selective allylic substitution of enantiopure allyl electrophiles with alkynyl Grignard reagent in 2009 [26-27], the first Cu-catalyzed asymmetric allylic substitution to achieve optically active 1,4-enynes from racemic substrates was reported by Hoveyda in 2011 (Scheme 2) [28]. By using NHC-Cu as the catalyst and alkynylaluminum reagent as the nucleophile generated in situ, a variety of chiral 1,4-enynes bearing all-carbon quaternary stereogenic centers were produced with modest to high yields, excellent regio- and enantioselectivities. Notably, α,β-unsaturated esters bearing a γ-phosphate group are suitable substrates under the established conditions, of which the products could be facilely converted into unsaturated chiral cyclic lactones through Au-catalyzed cyclization.

|
Download:
|
| Scheme 2. Asymmetric allylic substitution to achieve 1,4-enyne bearing all-carbon quaternary stereogenic centers. | |
{kind=link}
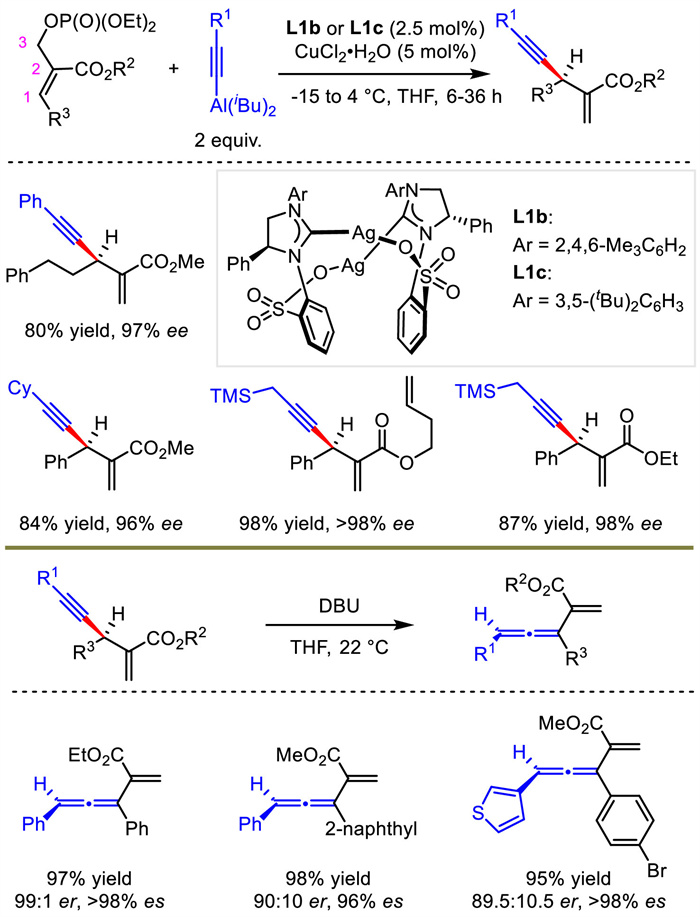
Later, Hoveyda's group extended the reaction scope to trisubstituted allylic phosphates containing a carboxylic ester group at C2 position with L1b or L1c as chiral ligands (Scheme 3) [8]. Under almost identical conditions, 1,4-enyne containing a tertiary stereogenic carbon center was efficiently prepared. The enantioenriched products were converted into chiral trisubstituted allenes through axial-to-central chirality transfer. A library of trisubstituted allenes with axial chirality were obtained with high ee and es values through this stereospecific proton migration approach, highlighting the potential application of this method in chemical synthesis.

|
Download:
|
| Scheme 3. Cu-catalyzed allylation with alkynylaluminum reagent and stereospecific proton migration to generate trisubstituted allene. | |
{kind=link}
In 2014, Sawamura and co-workers also described Cu/NHC-catalyzed allylic substitution reactions to yield enantioenriched 1,4-enynes [29,30]. Different from the necessity of sensitive alkynylaluminum reagent adopted in Hoveyda's work, which may be detrimental to the function group compatibility and operational convenience, terminal alkynes directly served as nucleophiles for this transformation. With Z-form allylic phosphate as the electrophile, the catalytic system displayed a broad substrate scope and high regio- and enantioselectivity control (Scheme 4). In contrast, the more general E-form allylic phosphates delivered products in very low yields and poor enantiocontrol. The authors proposed that the alkynyl copper(Ⅰ) intermediate would react with allylic electrophile via oxidative addition to generate Cu(Ⅲ) species and then underwent reductive elimination to provide the chiral 1,4-enynes.

|
Download:
|
| Scheme 4. Cu/NHC-catalyzed allylic alkynylation with terminal alkyne nucleophiles. | |
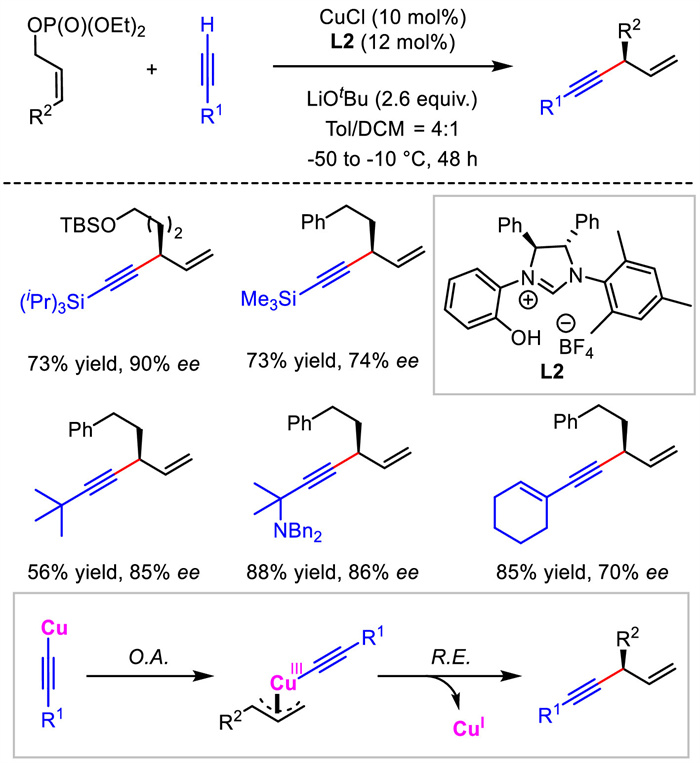{kind=link}
Different from NHC/Cu system developed by Hoveyda and Sawamura aforementioned, Tan and co-works described a chiral guanidine-ligated copper as the catalyst for allylic alkynylation in 2018 [31]. They used racemic cyclic allylic bromides as electrophiles and terminal alkynes as nucleophiles under a biphasic dichloromethane−water reaction system. Various 6-, 7- and 8-membered cyclic 1,4-enynes were obtained with high yields and good stereocontrol (Scheme 5).

|
Download:
|
| Scheme 5. (Guanidine) copper-catalyzed asymmetric allylic alkynylation. | |
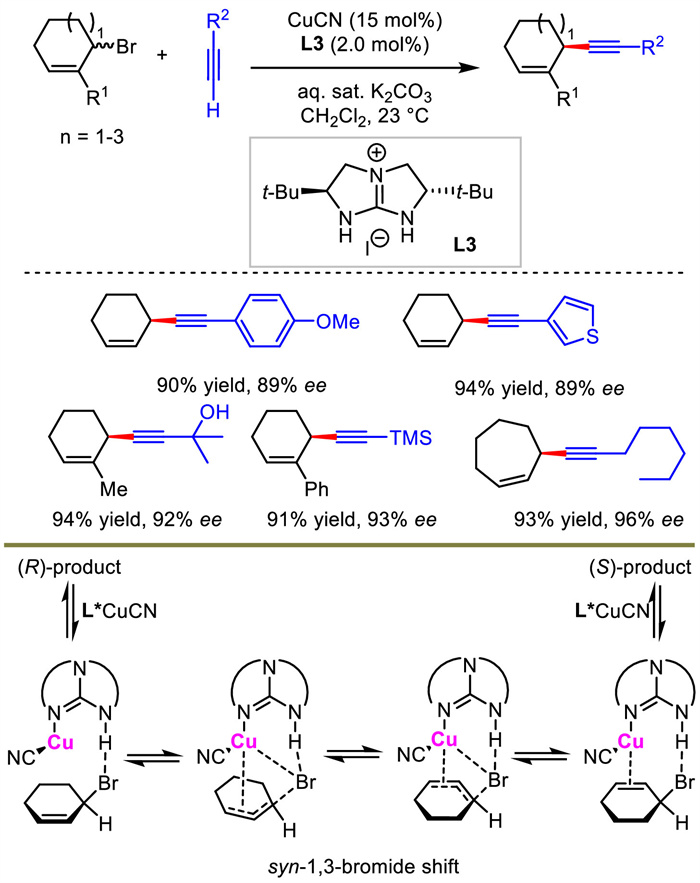{kind=link}
Interestingly, when enantioenriched allylic bromide was used as the substrate, under standard condition with no nucleophile, rapidly racemization was observed. In contrast, no racemization was detected in the absence of catalyst. Moreover, the author also found that allylic bromide always remains as a racemate in the whole course of standard reaction. According to these results together with DFT calculations, the author speculated that Cu(Ⅰ) catalyzed the racemization of allylic bromide with the assistance of intramolecular N-H···Br hydrogen bonding, which induced enantiomer interconversion equilibrium before nucleophilic attack, and thus realized a dynamic kinetic resolution process.
2.2. Ir catalysisIn 2013, Carreira's group reported the only example of Ir-catalyzed asymmetric allylic alkynylation to yield chiral 1,4-enynes (Scheme 6). Taking advantage of the Ir/(P, olefin) catalyst, substitutions of free allylic alcohols with alkynyl trifluoroborates proceeded in moderate to good yields, excellent regio- and enantioselectivities [32]. The reliability and applicability of this transformation were further highlighted by its application to the concise synthesis of AMG837, a biologically active molecule for the treatment of type Ⅱ diabetes. Different from previous alkynylmetal reagent or terminal alkynes serving as the nucleophiles, alkynyl trifluoroborates were adopted in present methodology and thus expanded the scope of reacting partners. Most recently, similar to L4, a 1,16-dihydroxytetraphenylene-derived (P, olefin)-ligand developed by Cui et al. also displayed high stereocontrol for this Ir-catalyzed allylic alkynylations [33].

|
Download:
|
| Scheme 6. Ir-catalyzed asymmetric allylic alkynylation and application. | |
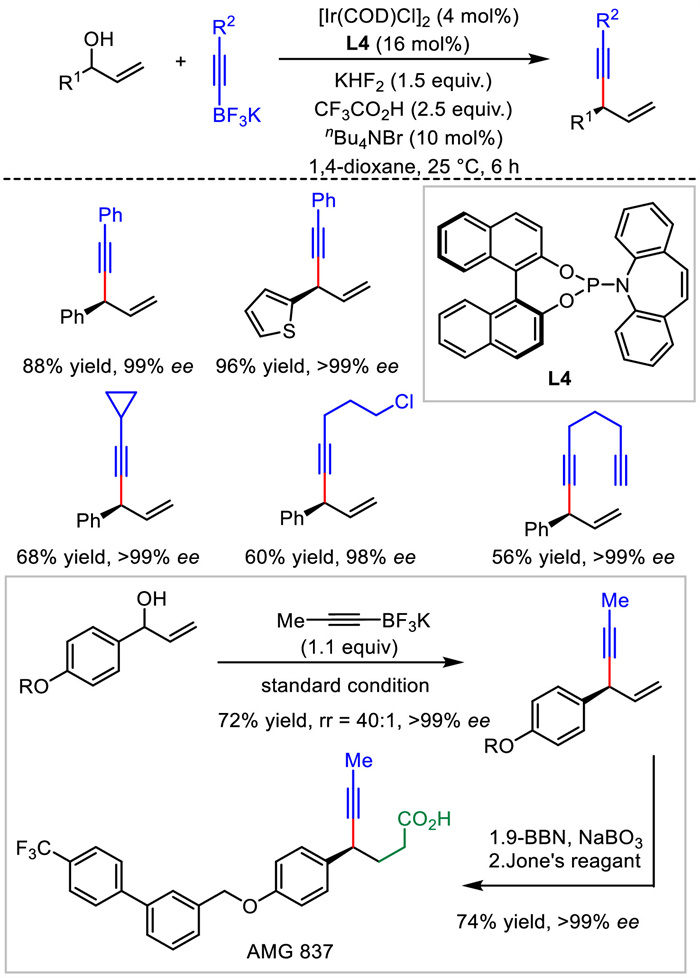{kind=link}
Obviously, these Ir-involved protocols were restricted to the use of aryl-substituted allyl alcohols as electrophiles. That is, alkyl-substituted allyl alcohols were not suitable for these transformations aforementioned. Soon after, an enantioselective alkynylation of alkyl-substituted allyl alcohols via kinetic resolution was reported by Zhou, Wong and Cui [34]. With the combination of iridium catalyst and tetraphenylene-derived (P, olefin)-ligand L5, a reasonable range of enantioenriched 1,4-enynes and alkyl allyl alcohols were obtained from racemic allyl alcohols with excellent enantioselectivities and with high s-factors (Scheme 7).

|
Download:
|
| Scheme 7. Ir-catalyzed enantioselective alkynylation of racemic allyl alcohols via kinetic resolution. | |
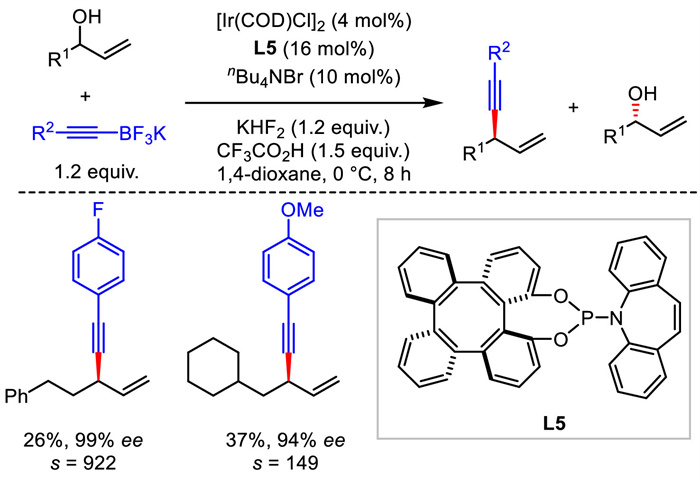{kind=link}
2.3. Rh catalysis
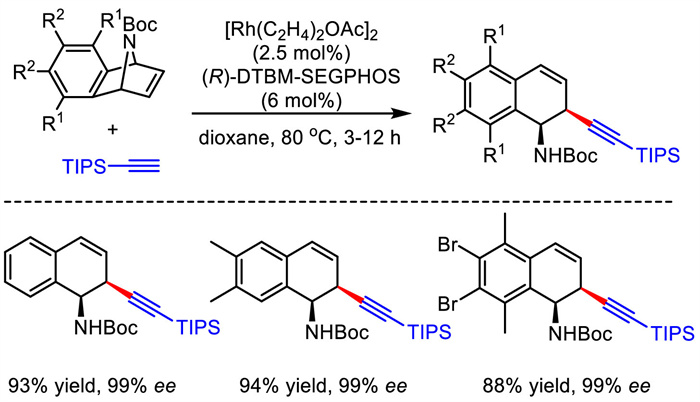
As early as 2008, Hayashi and Nishimura et al. realized enantioselective rhodium-catalyzed allylic alkynylation reaction [35]. They employed azabenzonorbornadienes as electrophiles and terminal alkynes as nucleophiles. The corresponding asymmetric desymmetrization proceeded smoothly under Rh/(R)-DTBM-SEGPHOS catalysis, and the ring-opening products were observed in high yields and excellent enantioselectivities (Scheme 8). The sterically bulky silyl substituent in the alkyne nucleophiles was necessary to guarantee the high enantioselectivity of the products.

|
Download:
|
| Scheme 8. Rh-catalyzed asymmetric desymmetrization with terminal alkynes. | |
{kind=link}
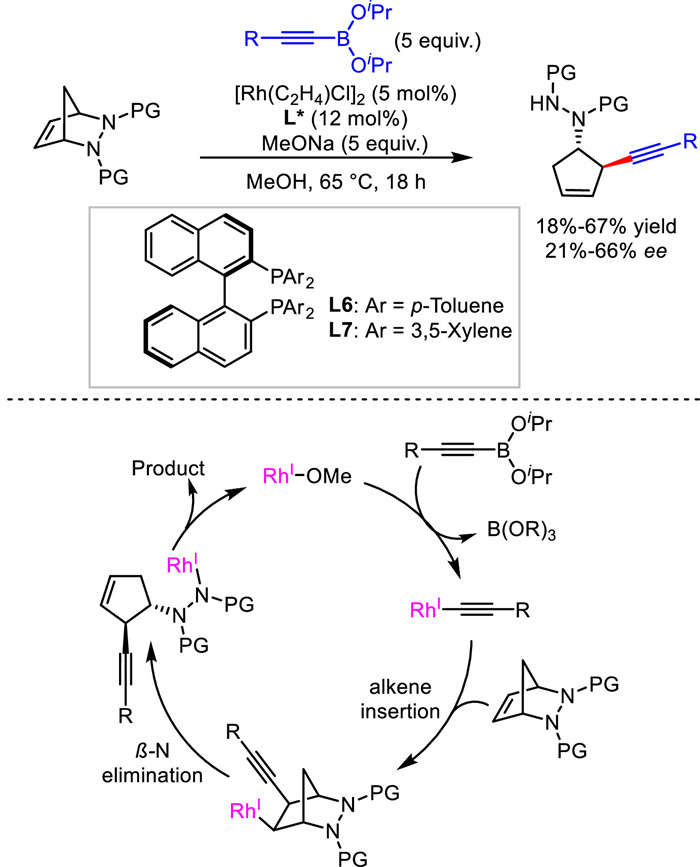
Similar to Hayashi's work, Pineschi's group also developed a desymmetrizative ring-opening reaction. Employing Rh/BINAP-type ligands as catalyst, alkynyl boronates as nucleophiles, a number of cyclic chiral 1,4-enynes were constructed from bicyclichydrazines but in moderate yields and enantioselectivities (Scheme 9) [36]. A proposed mechanism was outlined. Acetyliderhodium species from alkynylboronate reacted with alkene via migratory insertion to generate alkyl-Rh(Ⅰ) intermediate. Subsequent β-N elimination afforded the ring-opening product and regenerated Rh catalyst via protonation.

|
Download:
|
| Scheme 9. Rh-catalyzed asymmetric desymmetrization with terminal alkynyl boronates and proposed mechanism. | |
{kind=link}
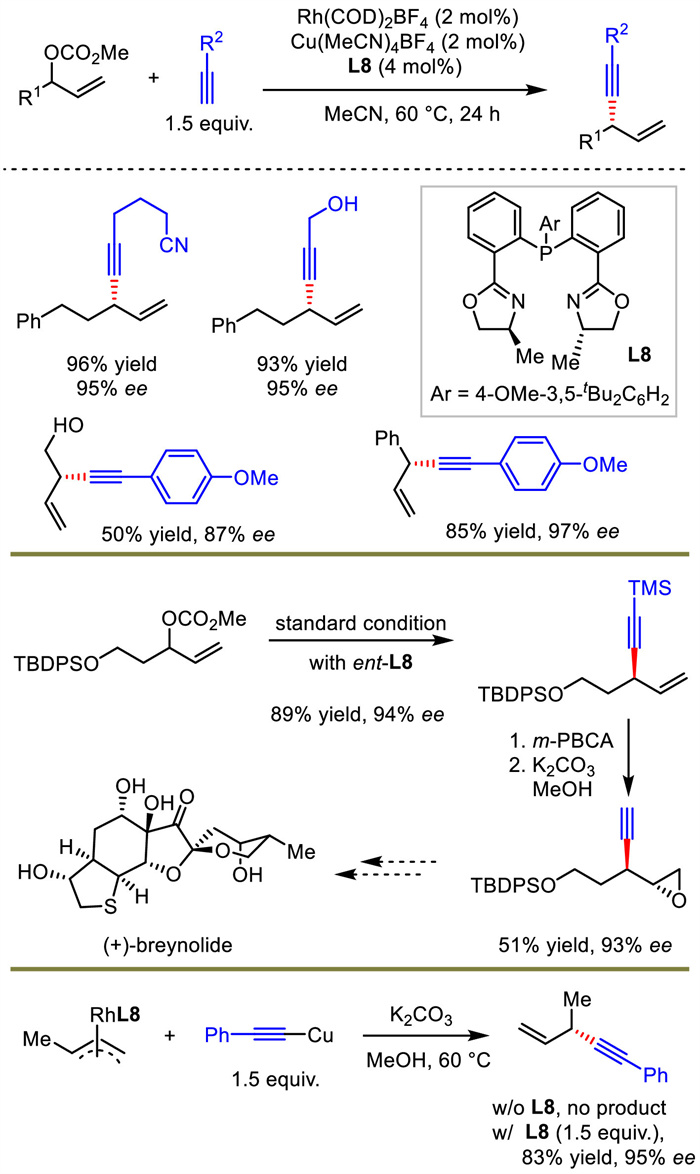
Considering the atom- and step-economy, undoubtedly, terminal alkynes should be the most direct and easily accessible nucleophiles for allylic alkynylation. Although Sawamura and Tan realized this goal, a lot of substrate limitations existed for allylic electrophiles, such as the requirement of Z-form or cyclic allylic partners [30,31]. Li et al. developed a bimetallic synergistic Rh/Cu catalysis system to realize the coupling of terminal alkynes and racemic allylic carbonates in 2020 (Scheme 10) [37]. This method performed efficiently across a broad scope of reaction partners. Quaternary stereogenic centers were also feasible with this protocol but in poor enantioselectivities. Li further applied the method to the construction of the key fragment of (+)-breynolide.

|
Download:
|
| Scheme 10. Rh/Cu-cocatalyzed asymmetric allylic alkynylation. | |
{kind=link}
In this work, the authors conducted a series of experiments to uncover the critical mechanistic features. When allyl RhL8 complex was reacted with Cu-acetylide, additional equivalent of L8 was necessary to promote the reaction. In addition, the absolute configuration of the product was the same as that in allyl RhL8 complex. Based on these facts, copper was proposed to be simply engaged in acetylide formation and subsequent transmetalation to rhodium, followed by reductive elimination to generate chiral 1,4-enynes. This was very different from classical outer-sphere allylic substitution process. Additional ligand for copper was mandatory, presumably due to the role of extra L8 in facilitating the breaking of Cu acetylide oligomers and generating the key Cu-L8 acetylide for transmetalation.
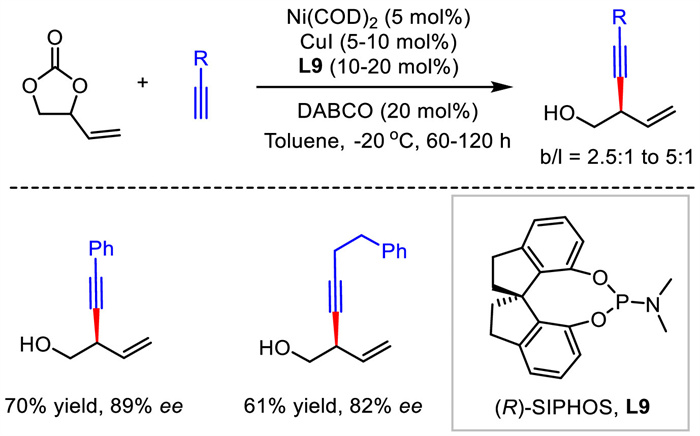
2.4. Ni catalysisIn 2021, Zhao described a Ni/Cu bimetallic catalytic system for the construction of enantioenriched 1,4-enynes (Scheme 11) [38]. With racemic vinyl ethylene carbonates (VEC) as electrophiles and terminal alkynes as nucleophiles, (R)-SIPHOS as the chiral ligand, a group of 1,4-enynes bearing a hydroxy unit were prepared with good enantioselectivities and moderate regiocontrol. DFT calculations suggested a transmetalation from Cu(Ⅰ)-acetylide to Ni(Ⅱ) which was generated from the oxidative addition of Ni(0) by VEC and subsequent reductive elimination process.

|
Download:
|
| Scheme 11. Ni/Cu-cocatalyzed asymmetric allylic alkynylation. | |
{kind=link}
2.5. Radical allylic C-H alkynylation
Previously, allylic alkynylation required the use of classical allylic electrophiles bearing suitable leaving groups. In contrast, a more straightforward alternative strategy should be direct allylic C-H alkynylation. Recently, Liu's group reported a copper-catalyzed asymmetric radical oxidative coupling between allylic C(sp3)-H bond and C(sp)-H bond of terminal alkynes (Scheme 12) [39]. Both acyclic and cyclic olefin electrophiles were suitable for accessing 1,4-enynes with moderate yields and moderate to good enantioselectivities. The reactions proceeded under mild conditions but needed a long reaction time. Although P(Ⅲ) ligand was adopted, it was thought to be oxidized to form P(Ⅴ) under oxidants. Thus, the key to the success of this scenario is the rationally designed anionic N, N, P(O) tridentate ligands featuring chiral oxazoline and pentavalent phosphine oxide which are resistant to the strong oxidative condition required for hydrogen atom abstraction.

|
Download:
|
| Scheme 12. Cu-catalyzed asymmetric radical allylic C-H alkynylation. | |
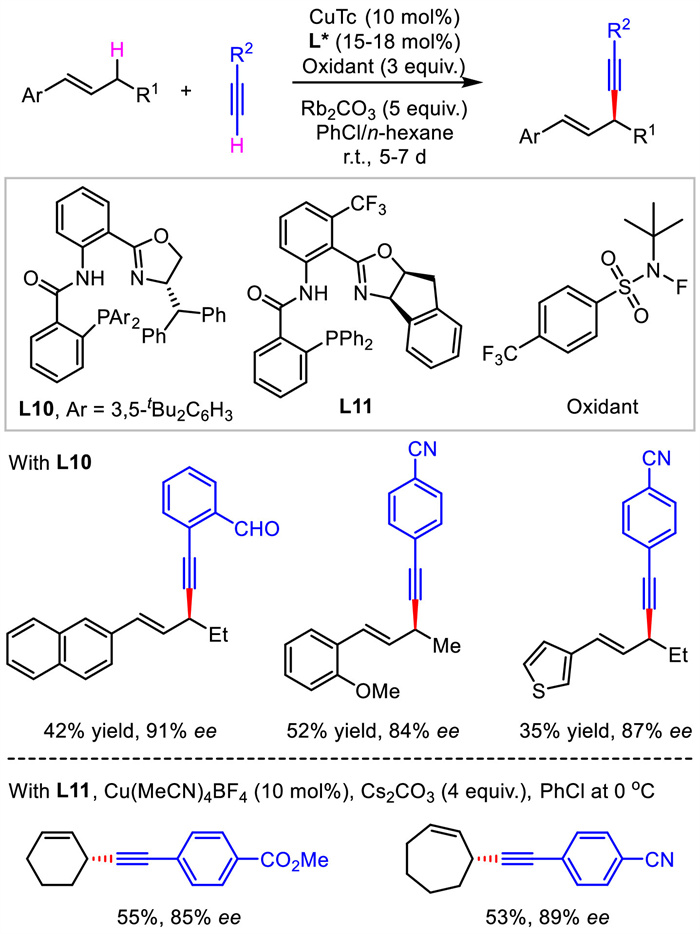{kind=link}
A plausible mechanism was also proposed (Scheme 13). First, the generated Cu(Ⅰ)-acetylide A would be oxidized to form Cu(Ⅱ) species B via a single electron transfer process. The resulting N-centered radical abstracted a hydrogen atom from allylic electrophile to generate allyl radical C which would then react with B to provide high-valent Cu(Ⅲ) intermediate D. Finally, a reductive elimination from D occurred to furnish the product and regenerate Cu(Ⅰ).

|
Download:
|
| Scheme 13. Proposed mechanism for Cu-catalyzed asymmetric radical allylic C-H alkynylation. | |
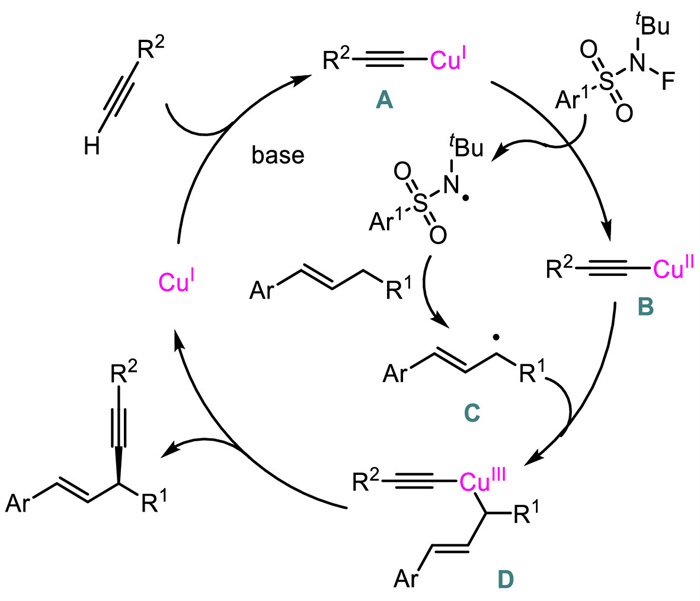{kind=link}
2.6. Organocatalytic allylic alkynylation
In 2016, Shibata et al. provided a metal-free protocol for asymmetric allylic alkynylation. With (DHQD)2PHAL as the organocatalyst, they exploited Baylis–Hillman type reaction between unsaturated ester containing an allylic fluoride leaving group and TMS-substituted alkyne (Scheme 14) [40]. A variety of corresponding 1,4-enyne products were obtained in moderate to good enantioselectivities. The chair-like Zimmerman−Traxler transition state was proposed to explain the observed stereocontrol outcome. The simultaneous Si-F and Lewis base-promoted allylation interactions are thought to be responsible for the activation of C-F bond. Thus, no fully charged species was generated, which inhibited the possible racemization of products under basic condition.

|
Download:
|
| Scheme 14. Organocatalytic asymmetric allylic alkynylation and proposed transition state. | |
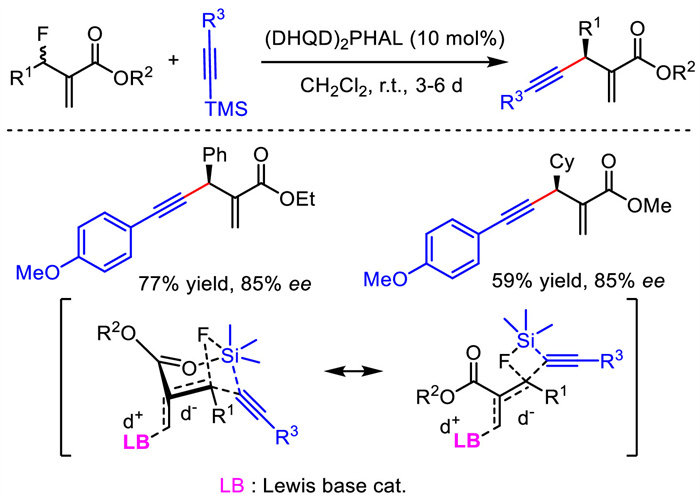{kind=link}
3. Asymmetric propargylic substitution with alkenyl nucleophiles 3.1. Cu catalysis
Compared with asymmetric allylic alkynylation, the asymmetric propargylic olefination was less developed. A decade ago, Maarseveen and Nishibayashi sequentially reported one related case independently in low stereocontrol via copper-catalyzed propargylic amination (Scheme 15) [41,42]. The reactions were proposed to undergo the formation of critical copper-allenylidene intermediate.

|
Download:
|
| Scheme 15. Two cases for Cu-catalyzed asymmetric propargylation for the synthesis of 1,4-enynes. | |
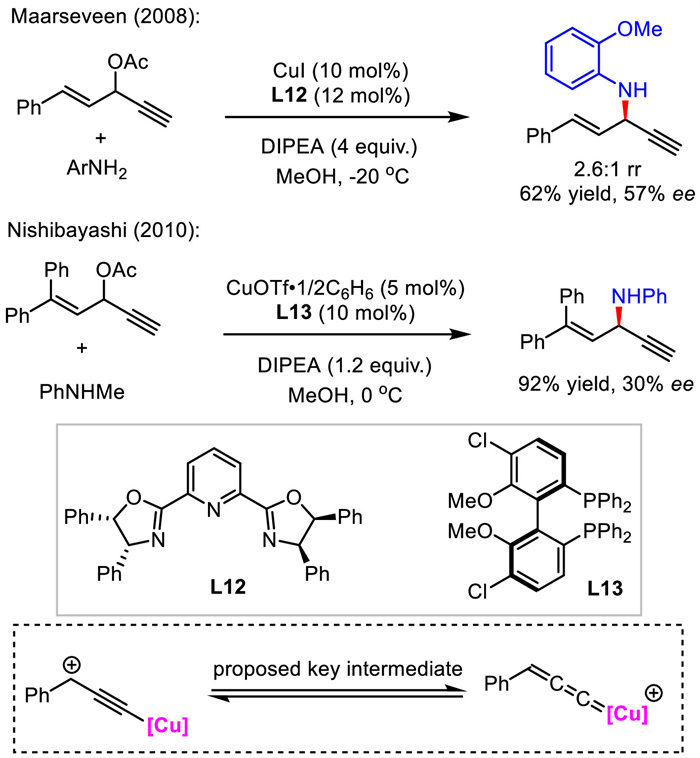{kind=link}
Until 2017, Xiao's group described a novel asymmetric propargylation of propargylic acetates with phosphorus ylides [43]. The newly generated phosphorus ylide products could be quenched by Wittig reaction with aldehyde or ketene in a one-pot sequence. Thus, various 1,4-enynes and 1,4-allenynes bearing different function groups were obtained with remarkable yields and stereoselectivities (Scheme 16). The phosphorus ylide substrate was used as a precursor of olefin and allene and might find additional applications in other reaction systems. Similar copper-allenylidene intermediate was proposed in the catalytic cycle in this work.

|
Download:
|
| Scheme 16. Cu-catalyzed asymmetric propargylic substitution cascade with Wittig reaction. | |
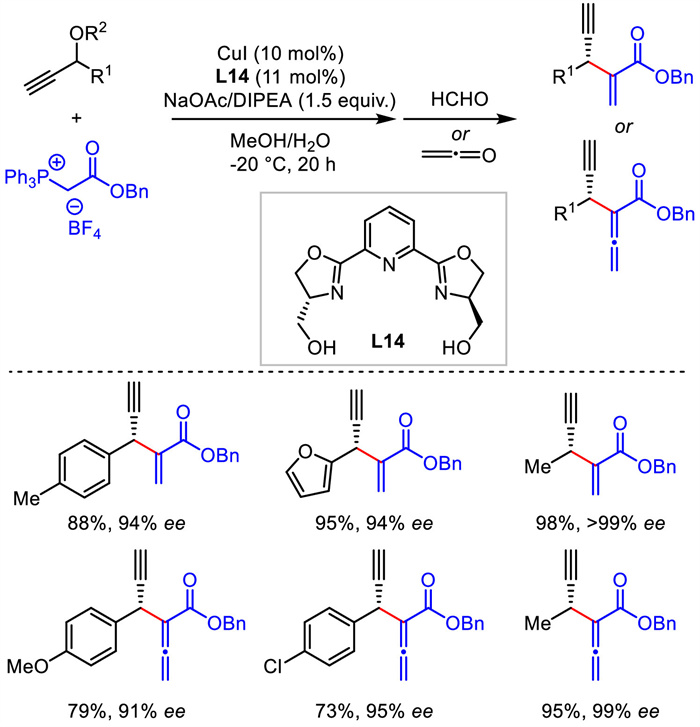{kind=link}
Although the construction of chiral 1,4-enynes from 1,4-enyne electrophiles via Cu-catalyzed propargylation was shown to be challenging [41,42], Gong and Song realized this process via a synergistic Cu/N-heterocyclic carbene catalysis in 2022 [44]. With 1,4-enyne bearing an internal leaving group under copper catalyst as the electrophile and aldehyde under NHC as the nucleophile, a diastereo- and enantio-selective coupling performed smoothly. Generally, high stereocontrol was observed but with a low yield (Scheme 17).

|
Download:
|
| Scheme 17. Cu-catalyzed asymmetric propargylic substitution of 1,4-enynes. | |
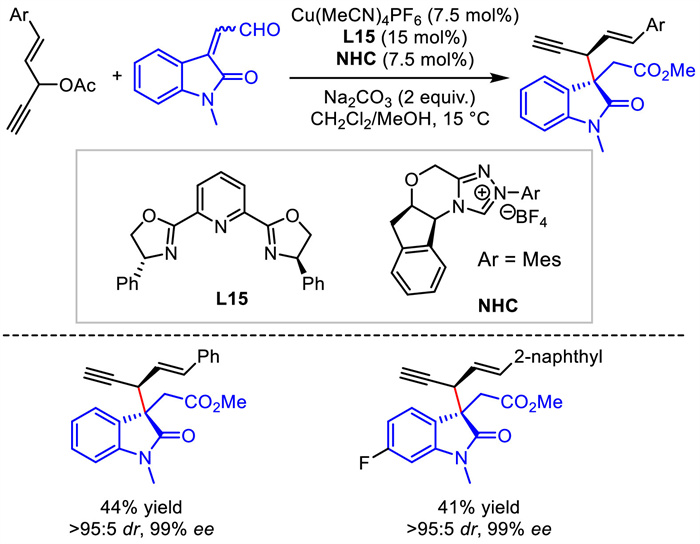{kind=link}
Recently, Liu's group described a copper-catalyzed asymmetric propargylic substitution of propargyl bromides with alkenylboronate esters [45]. With a novel cinchona alkaloid-derived hemilabile N,N,N-tridentate ligand, remarkable enantioselectivities were generally obtained for the 1,4-enyne products. From the potential mechanism, the reaction was proposed to follow an enantioconvergent radical cross-coupling pathway, in which alkenyl Cu(Ⅰ) was oxidized by propargylic bromide to generate alkenyl Cu(Ⅱ) species and propargylic carbon radical. The subsequent coupling via a possible reductive elimination from Cu(Ⅲ) intermediate provided the final product (Scheme 18).

|
Download:
|
| Scheme 18. Copper-catalyzed asymmetric propargylic substitution with alkenyl bromide. | |
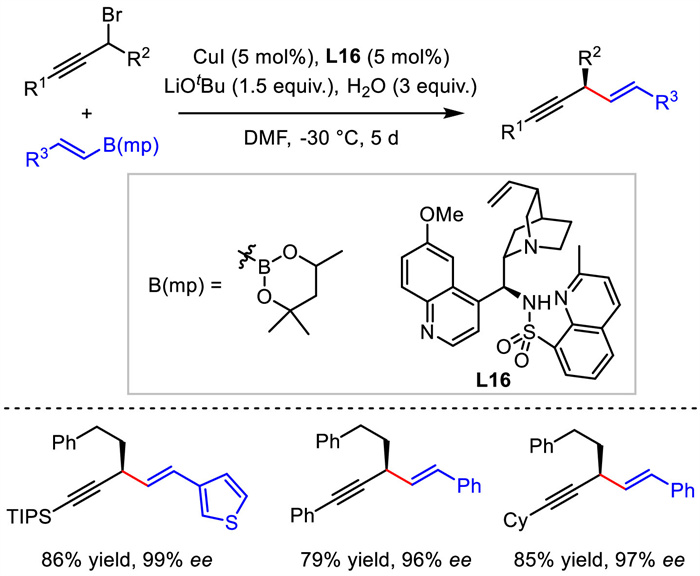{kind=link}
3.2. Organocatalytic propargylation
The only organocatalytic propargylation was published by Maruoka group in 2019. With the employment of a highly acidic chiral N-triflyl phosphoramide as Brønsted acid catalyst and alkenyl boroxine as reaction partner, free propargylic alcohols containing diverse functional groups were smoothly alkenylated in good to excellent enantioselectivities (Scheme 19) [46]. Both aryl and alkyl-substituted propargylic alcohols were suitable for the transformation. A plausible transition state was given. The Brønsted acid played the activation via hydrogen-bonding interaction with the electrophile and electron-donating effect with the nucleophile.

|
Download:
|
| Scheme 19. Organocatalytic asymmetric propargylic substitution with alkenyl boroxine. | |
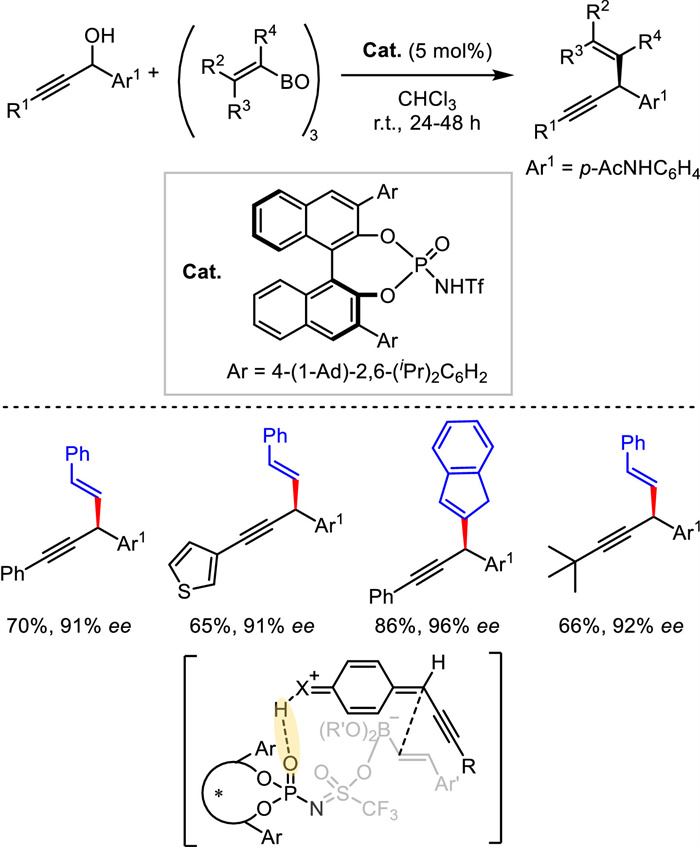{kind=link}
4. Asymmetric alkynylallylic substitution 4.1. Mo catalysis
Different from allylic substitution and propargylic substitution strategies, alkynylallylic substitution uses 1,3-enyne bearing an allylic leaving group as the electrophilic substrate. It provides an alternative but more direct approach to 1,4-enynes. Early in 1999, Trost et al. disclosed molybdenum-catalyzed asymmetric alkynylallylic substitution with stabilized carbon nucleophiles, furnishing 1,4-enynes with excellent enantioselectivities and moderate regioselectivities (Scheme 20) [47]. This is the first example of asymmetric alkynylallylic substitution and the only example of molybdenum-mediated process up to now.

|
Download:
|
| Scheme 20. Alkynylallylic substitution via Mo catalyst. | |
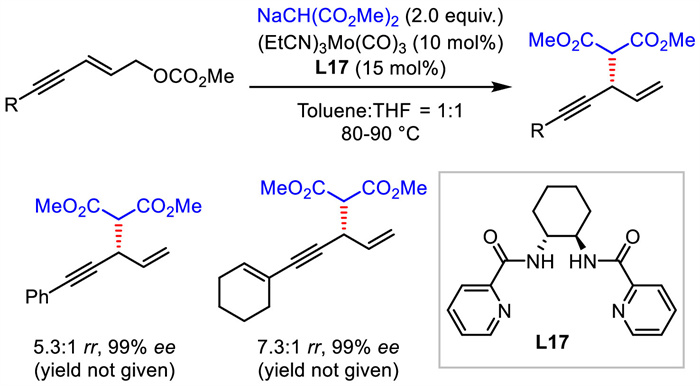{kind=link}
4.2. Cu catalysis
In 2004, Hoveyda's group established Cu/dipeptide ligand-catalyzed asymmetric alkynylallylic alkylation reaction with alkylzinc reagent as the nucleophile (Scheme 21) [48]. Two 1,4-enynes were obtained in good yields and enantioselectivities. Especially, a quaternary stereogenic carbon center was also smoothly constructed via this protocol. A Cu(Ⅲ)-involved mechanistic process was generally proposed for this type of reactions.

|
Download:
|
| Scheme 21. Redox-cyclic Cu-catalyzed asymmetric alkynylallylic substitutions to generate 1,4-enynes. | |
{kind=link}
A general approach for copper-catalyzed asymmetric alkynylallylic substitution was documented by Alexakis group in 2012. Instead of zinc nucleophile, Alexakis utilized more readily available Grignard reagent as the alkylation nucleophile coupling with alkynylallylic chloride as the electrophile (Scheme 21) [49]. With copper and phosphoramidite L19 as the catalyst, the transformation generally exhibited good reactivity, regioselectivity and enantioselectivity with a series of alkyl Grignard reagents. However, when phenyl Grignard reagent was used, substitution via SN2 pathway was observed as the only product. In addition, in the case of methyl magnesium bromide, nearly no regioselectivity was observed, leading to a nearly 1:1 mixture of branch/linear regioisomers.
In 2022, He's group communicated a general protocol for redox-free Cu-catalyzed asymmetric alkynylallylic substitution [50]. They demonstrated that both Cu(Ⅰ) and Cu(Ⅱ) could catalyze the transformation in high yield and excellent regio- and enantioselectivity. Remarkably, the reaction underwent smoothly in gram-scale even under water and air, demonstrating the practicability and robustness of the process. A variety of functional groups on amine nucleophiles and 1,3-enyne electrophiles showed high compatibility under mild and robust conditions. Diverse bioactive molecules derived substrates were suitable for the substitutions (Scheme 22).

|
Download:
|
| Scheme 22. Redox-free Cu-catalyzed asymmetric alkynylallylic substitutions. | |
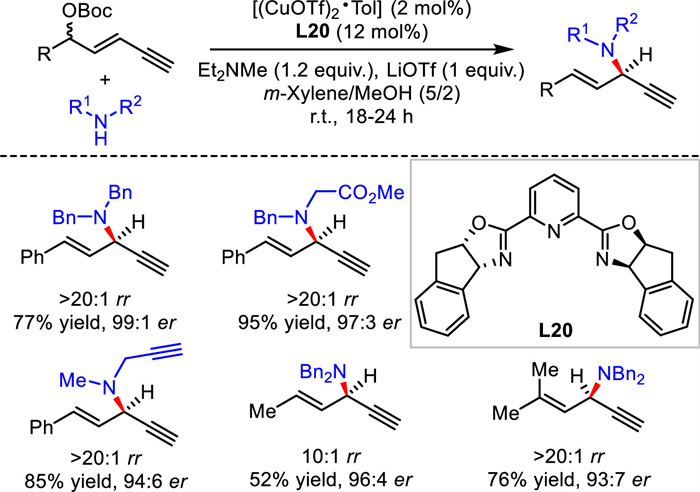{kind=link}
In addition to alkynylallylic amination, the present protocols were further extended to cover asymmetric alkynylallylic alkoxylation and alkylation with alcohol and enol as nucleophiles respectively (Scheme 23). Both catalytic systems relied on the combination of copper catalyst and boronic acid as a cocatalyst. Therefore, alkynylallylic alkoxylation and alkylation proceeded well and good yields and stereoselectivities were obtained. At the same time, an intramolecular decarboxylative alkynylallylic substitution process was also disclosed by He's group (Scheme 23). Similarly, high reaction efficiency and stereocontrol were observed with different substrates. It provides an alternative pathway for the intermolecular process aforementioned. This catalytic system was also very effective for classical propargylic substitutions that failed to construct 1,4-enynes in high stereocontrol or efficiency [41,42,44]. Based on this result, process, regio- and enantioconvergent transformation was carried out and provided a single product from eight substrates.

|
Download:
|
| Scheme 23. Intermolecular alkynylallylic alkoxylation and alkylation and decarboxylative alkynylallylic substitution. | |
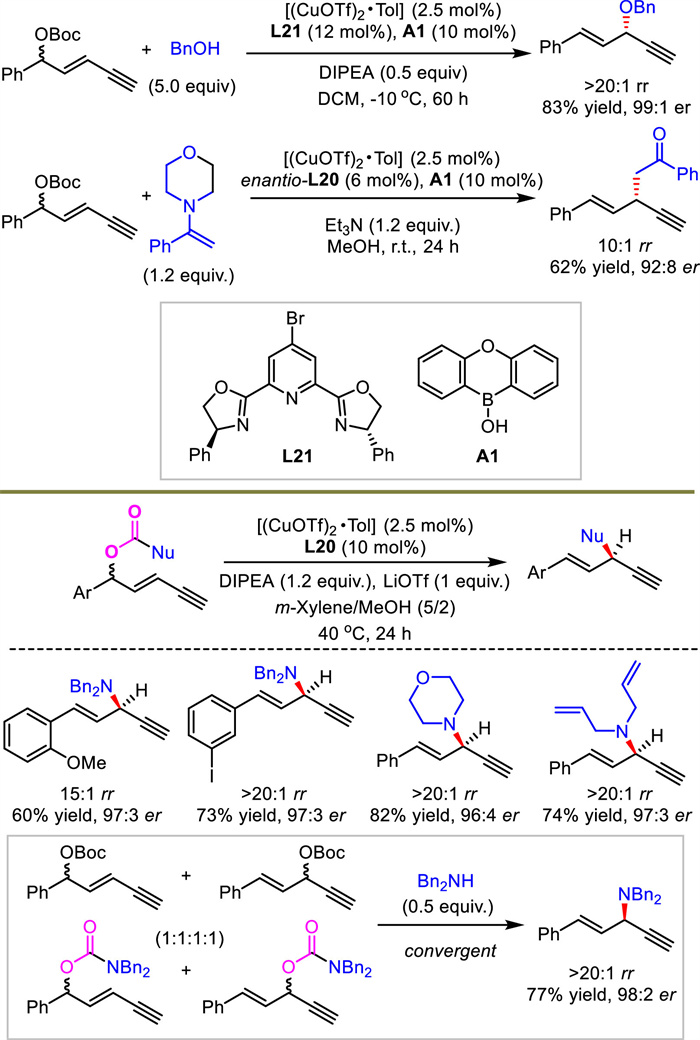{kind=link}
The authors conducted a series of mechanistic experiments to elucidate the possible mechanism (Scheme 24). Comparing the X-ray structures of Cu(Ⅰ) and Cu(Ⅱ), they found that the latter featured a mononuclear metal center ligated with two chiral ligands, different from classical Cu(Ⅰ) complex possessing dinuclear metal centers with two chiral ligands. Kinetic studies uncovered that the reaction was first order on catalyst but zero order on both two substrates. Finally, a proposed mechanism was described. Copper first reacted with 1,3-enyne under base to give alkynyl copper intermediate Int-1, which then generated the critical Cu-allenylidene species Int-3. A following outer-sphere nucleophilic substitution and proton transfer released the product and catalyst. The formation of Int-3 was the rate-limiting step for the catalytic cycle.

|
Download:
|
| Scheme 24. Mechanistic studies and proposed mechanism for Cu-catalyzed redox-free alkynylallylic substitutions. | |
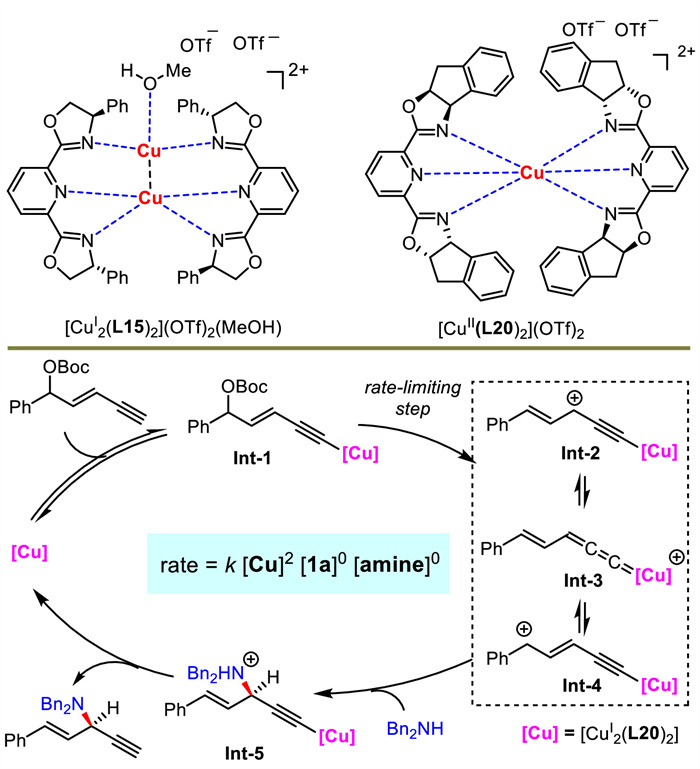{kind=link}
5. Asymmetric hydroalkynylation 5.1. Ni-catalyzed process with diene
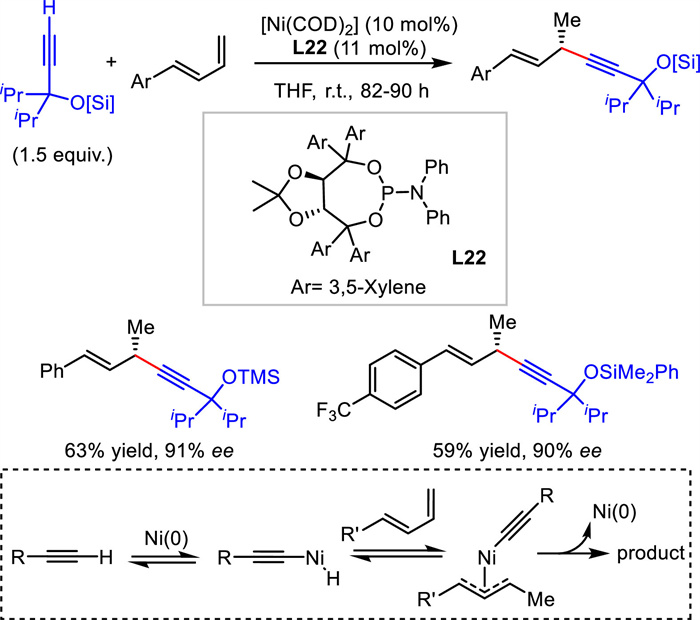
Different from classical substitutions with the requirement of a prestored leaving group in the electrophile, hydroalkynylation of unsaturated bond features 100% atom economy and convenient availability of substrates. In 2010, Suginome and coworkers reported the first Ni-catalyzed asymmetric hydroalkynylation of dienes. With TADDOL-derived phosphoramidite L22 and Ni(0) as the catalyst, 1,4-enynes were achieved with > 90% ee values (Scheme 25) [51]. A possible mechanism [52] involved the oxidation of terminal alkyne to Ni(0) to generate alkynyl Ni(Ⅱ) species, which would insert into diene to form a thermodynamic stable allylic Ni intermediate. A final reductive elimination process provided the product and regenerated Ni(0).

|
Download:
|
| Scheme 25. Ni-catalyzed asymmetric hydroalkynylation of dienes. | |
{kind=link}
5.2. Rh-catalyzed decarboxylative process with allene
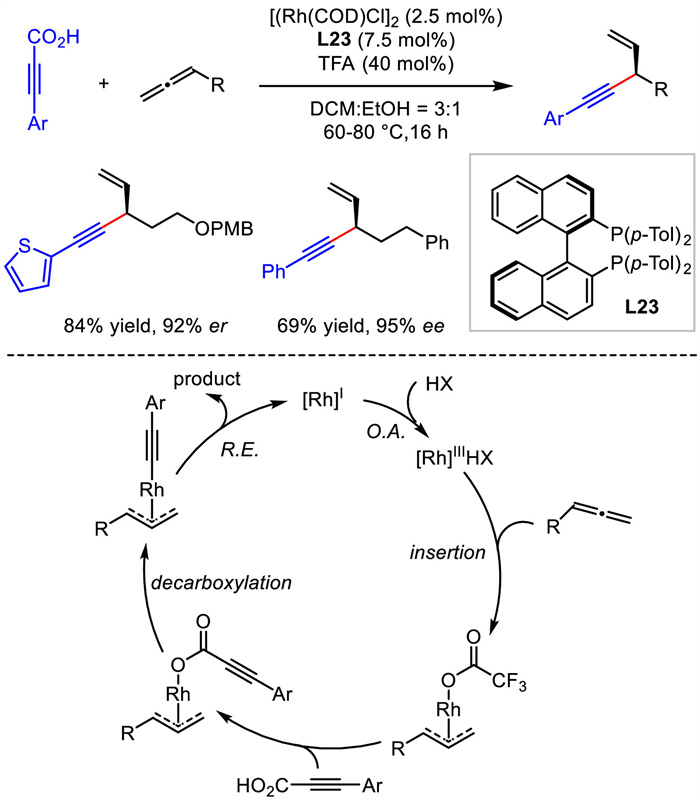
In 2018, Breit disclosed a rhodium catalyzed asymmetric hydroalkynylation of allenes. Instead of typical terminal alkynes used as nucleophiles, they exploited arylpropiolic acids as alkyne surrogates. A broad scope of allenes and propiolic acids readily underwent this reaction to deliver corresponding compounds with good yields and enantioselectivities (Scheme 26) [53].

|
Download:
|
| Scheme 26. Rh-catalyzed decarboxylative hydroalkynylation of allenes. | |
{kind=link}
Preliminary studies indicated the existence of interconvertible π -σ -π allylrhodium intermediate. The crossover experiments implied that allylic alkynyl ester was kind of off-cycle product. A plausible catalytic pathway was proposed. Initially, a Rh(Ⅲ) hydride complex was formed via oxidative addition of Rh(Ⅰ) by trifluoroacetic acid. The insertion of allene by Rh(Ⅲ) hydride provided η3-allylrhodium species and η1-allylrhodium species which could convert to each other through π −σ −π isomerization. After the ligand exchange with arylpropiolic acid and following decarboxylation, allyl alkynyl rhodium species was formed. Ultimately, reductive elimination from this species yielded the product and regenerated Rh catalyst.
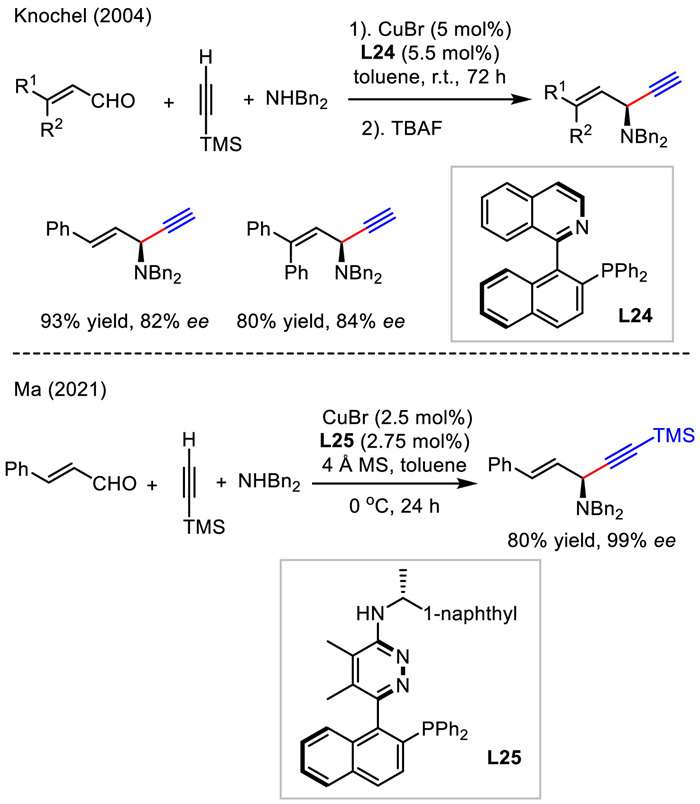
6. Asymmetric 1,2-addition of alkynes to conjugated aldehydes and iminesThree-component coupling of aldehyde, alkyne, and amine (A3 coupling) is an attractive and efficient pathway for rapid construction of complex skeletons. Nevertheless, a general catalytic asymmetric method for the use of conjugated aldehydes or imines has remained elusive for a long time. In 2004, Knochel reported two cases for Cu-catalyzed asymmetric A3 coupling of unsaturated aldehyde, terminal alkyne and secondary amine (Scheme 27) [54]. High yields and good enantioselectivities were observed for 1,4-enyne products with the use of copper catalyst and L24. Recently, Ma's group modified ligand L24 and obtained a new ligand L25 which showed much higher enantiocontrol than that in Knochel's work (99% vs. 82%). This reaction also featured lower temperature and shorter reaction time [55].

|
Download:
|
| Scheme 27. Cu-catalyzed asymmetric 1,2-addition of alkynes to conjugated imines. | |
{kind=link}
In addition, aldehydes are known to be less electrophilic than imines. With the use of chiral Lewis acid generated in situ, Pu's group developed several systems to realize this type of transformation [56-58]. However, a high chiral ligand loading (20%–40%) and equivalent of transition metals were generally required. Similarly, Xu designed new chiral β-hydroxy amide ligands for this reaction and observed good enantiocontrol with high catalyst loading [59].
7. ConclusionsThe present review provides an overview of the hitherto advances that have been made in catalytic asymmetric synthesis of 1,4-enynes. Elegant methods divided into five major strategies have been depicted. Comparing with the extensive studies and applications of allyl and propargyl motifs bearing stereogenic centers, chiral 1,4-enynes have received much less attention. Considering the much higher application potential of 1,4-enynes via controllable modifications of both alkenyl and alkynyl units, novel and efficient synthetic protocols for the construction of 1,4-enynes are still heavily needed. In especial, several obvious limitations exist. First, with respect to molecular diversity, the present protocols are limited to asymmetric construction of C-C, C-N and C-O bonds. To expand the product diversity and application space, approaches towards chiral 1,4-enynes bearing other heteroatom-containing stereocenters including S, P, B, Si, represent intriguing direction. Meanwhile, methods for the construction of quaternary stereocenters in 1,4-enynes are limited. In addition, asymmetric alkynylallylic substitution represents a direct route to achieve 1,4-enynes and has displayed high efficiency and generality, but still remains underdeveloped. We hope this short review will provide a useful summary and perspective for the synthetic community and inspire further exciting developments in this rapidly evolving research area.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (NSFC, No. 22071262), the Science and Technology Commission of Shanghai Municipality (No. 22ZR1475200) and Shanghai Rising-Star program (No. 20QA1411300).
| [1] |
M. Miyazawa, H. Shimamura, R.C. Bhuva, S.I. Nakamura, H. Kameoka, J. Agric. Food Chem. 44 (1996) 3444-3448. DOI:10.1021/jf960025a |
| [2] |
M. Hayashi, H. Miyake, S. Kori, et al., J. Med. Chem. 23 (1980) 519-524. DOI:10.1021/jm00179a010 |
| [3] |
G. Stork, M. Isobe, J. Am. Chem. Soc. 97 (1975) 4745-4746. DOI:10.1021/ja00849a042 |
| [4] |
K. Iida, M. Hirama, J. Am. Chem. Soc. 116 (1994) 10310-10311. DOI:10.1021/ja00101a065 |
| [5] |
T.P. Heffron, T.F. Jamison, Org. Lett. 5 (2003) 2339-2342. DOI:10.1021/ol0347040 |
| [6] |
V. Hickmann, M. Alcarazo, A. Furstner, J. Am. Chem. Soc. 132 (2010) 11042-11044. DOI:10.1021/ja104796a |
| [7] |
R. Bajpai, D.P. Curran, J. Am. Chem. Soc. 133 (2011) 20435-20443. DOI:10.1021/ja2082679 |
| [8] |
J.A. Dabrowski, F. Haeffner, A.H. Hoveyda, Angew. Chem. Int. Ed 52 (2013) 7694-7699. DOI:10.1002/anie.201303501 |
| [9] |
X.F. Wei, T. Wakaki, T. Itoh, et al., Chem 5 (2019) 585-599. DOI:10.1016/j.chempr.2018.11.022 |
| [10] |
T. Fukuyama, Y. Ohta, C. Brancour, et al., Chem. Eur. J. 18 (2012) 7243-7247. DOI:10.1002/chem.201200045 |
| [11] |
S.A. Blaszczyk, D.A. Glazier, W. Tang, Acc. Chem. Res. 53 (2019) 231-243. |
| [12] |
X.N. Ke, C.M. Schienebeck, C.C. Zhou, X. Xu, W. Tang, Chin. Chem. Lett. 26 (2015) 730-734. DOI:10.1016/j.cclet.2015.03.016 |
| [13] |
D. Vasu, A. Das, R.S. Liu, Chem. Commun. 46 (2010) 4115-4117. DOI:10.1039/c0cc00071j |
| [14] |
G.Q. Chen, M. Shi, Chem. Commun. 49 (2013) 698-700. DOI:10.1039/C2CC37587G |
| [15] |
M. Shi, X.Y. Tang, G.Q. Chen, Synlett 25 (2014) 2311-2315. DOI:10.1055/s-0034-1378633 |
| [16] |
X.F. Wei, X.W. Xie, Y. Shimizu, M. Kanai, J. Am. Chem. Soc. 139 (2017) 4647-4650. DOI:10.1021/jacs.7b01254 |
| [17] |
S. Gao, H. Liu, C. Yang, et al., Org. Lett. 19 (2017) 4710-4713. DOI:10.1021/acs.orglett.7b01960 |
| [18] |
X. Fang, Q. Li, R. Shi, H. Yao, A. Lin, Org. Lett. 20 (2018) 6084-6088. DOI:10.1021/acs.orglett.8b02481 |
| [19] |
X. Chen, C.A. Baratay, M.E. Mark, X. Xu, P.W.H. Chan, Org. Lett. 22 (2020) 2849-2853. DOI:10.1021/acs.orglett.0c00929 |
| [20] |
C. Wu, W.J. Teo, S. Ge, ACS Catal. 8 (2018) 5896-5900. DOI:10.1021/acscatal.8b01410 |
| [21] |
N.A. Butt, W. Zhang, Chem. Soc. Rev. 44 (2015) 7929-7967. DOI:10.1039/C5CS00144G |
| [22] |
L. Süsse, B.M. Stoltz, Chem. Rev. 121 (2021) 4084-4099. DOI:10.1021/acs.chemrev.0c01115 |
| [23] |
O. Pàmies, J. Margalef, S. Cañellas, et al., Chem. Rev. 121 (2021) 4373-4505. DOI:10.1021/acs.chemrev.0c00736 |
| [24] |
M. Liu, H. Zhao, C. Li, Chin. Chem. Lett. 32 (2021) 385-388. DOI:10.1016/j.cclet.2020.04.009 |
| [25] |
Z.L. Zhao, Q. Gu, X.Y. Wu, S.L. You, Chin. Chem. Lett. 27 (2016) 619-622. DOI:10.1016/j.cclet.2016.02.017 |
| [26] |
Y. Kiyotsuka, Y. Kobayashi, J. Org. Chem. 74 (2009) 7489-7495. DOI:10.1021/jo901728b |
| [27] |
Q. Wang, Y. Kobayashi, Tetrahedron Lett. 51 (2010) 5592-5595. DOI:10.1016/j.tetlet.2010.08.051 |
| [28] |
J.A. Dabrowski, F. Gao, A.H. Hoveyda, J. Am. Chem. Soc. 133 (2011) 4778-4781. DOI:10.1021/ja2010829 |
| [29] |
Y. Makida, Y. Takayama, H. Ohmiya, M. Sawamura, Angew. Chem. Int. Ed. 52 (2013) 5350-5354. DOI:10.1002/anie.201300785 |
| [30] |
A. Harada, Y. Makida, T. Sato, H. Ohmiya, M. Sawamura, J. Am. Chem. Soc. 136 (2014) 13932-13939. DOI:10.1021/ja5084333 |
| [31] |
X.Y. Cui, Y. Ge, S.M. Tan, et al., J. Am. Chem. Soc. 140 (2018) 8448-8455. DOI:10.1021/jacs.7b12806 |
| [32] |
J.Y. Hamilton, D. Sarlah, E.M. Carreira, Angew. Chem. Int. Ed. 52 (2013) 7532-7535. DOI:10.1002/anie.201302731 |
| [33] |
J. Guo, W.B. Xiong, H.R. Ma, et al., Chem. Sci. 13 (2022) 4608-4615. DOI:10.1039/d2sc00388k |
| [34] |
J. Guo, H.R. Ma, W.B. Xiong, et al., Chem. Sci. 13 (2022) 13914-13921. DOI:10.1039/d2sc04892b |
| [35] |
T. Nishimura, E. Tsurumaki, T. Kawamoto, X.X. Guo, T. Hayashi, Org. Lett. 10 (2008) 4057-4060. DOI:10.1021/ol801549t |
| [36] |
S. Crotti, F. Bertolini, F. Macchia, M. Pineschi, Chem. Commun. 2008 (2008) 3127-3129. DOI:10.1039/b803693d |
| [37] |
W.Y. Huang, C.H. Lu, S. Ghorai, B. Li, C. Li, J. Am. Chem. Soc. 142 (2020) 15276-15281. DOI:10.1021/jacs.0c08283 |
| [38] |
Y. Huang, C. Ma, S. Liu, et al., Chem 7 (2021) 812-826. DOI:10.1016/j.chempr.2021.02.018 |
| [39] |
L. Liu, K.X. Guo, Y. Tian, et al., Angew. Chem. Int. Ed. 60 (2021) 26710-26717. DOI:10.1002/anie.202110233 |
| [40] |
S. Okusu, H. Okazaki, E. Tokunaga, V.A. Soloshonok, N. Shibata, Angew. Chem. Int. Ed. 55 (2016) 6744-6748. DOI:10.1002/anie.201601928 |
| [41] |
R.J. Detz, M.M. Delville, H. Hiemstra, J.H. van Maarseveen, Angew. Chem. Int. Ed. 47 (2008) 3777-3780. DOI:10.1002/anie.200705264 |
| [42] |
G. Hattori, K. Sakata, H. Matsuzawa, et al., J. Am. Chem. Soc. 132 (2010) 10592-10608. DOI:10.1021/ja1047494 |
| [43] |
K. Zhang, L.Q. Lu, S. Yao, et al., J. Am. Chem. Soc. 139 (2017) 12847-12854. DOI:10.1021/jacs.7b08207 |
| [44] |
Y.H. Wen, Z.J. Zhang, J.Song S.Li, L.Z. Gong, Nat. Commun. 13 (2022) 1344-1353. DOI:10.1038/s41467-022-29059-0 |
| [45] |
P.F. Wang, J. Yu, K.X. Guo, et al., J. Am. Chem. Soc. 144 (2022) 6442-6452. DOI:10.1021/jacs.2c00957 |
| [46] |
J.F. Bai, K. Yasumoto, T. Kano, K. Maruoka, Angew. Chem. Int. Ed. 58 (2019) 8898-8901. DOI:10.1002/anie.201904520 |
| [47] |
B.M. Trost, S. Hildbrand, K. Dogra, J. Am. Chem. Soc. 121 (1999) 10416-10417. DOI:10.1021/ja992602s |
| [48] |
M.A. Kacprzynski, A.H. Hoveyda, J. Am. Chem. Soc. 126 (2004) 10676-10681. DOI:10.1021/ja0478779 |
| [49] |
H. Li, A. Alexakis, Angew. Chem. Int. Ed. 51 (2012) 1055-1058. DOI:10.1002/anie.201107129 |
| [50] |
J.S. Ma, H.Y. Lu, Y.W. Chen, et al., Nat. Synth. 2 (2023) 37-48. |
| [51] |
M. Shirakura, M. Suginome, Angew. Chem. Int. Ed. 49 (2010) 3827-3829. DOI:10.1002/anie.201001188 |
| [52] |
M. Shirakura, M. Suginome, J. Am. Chem. Soc. 130 (2008) 5410-5411. DOI:10.1021/ja800997j |
| [53] |
C.P. Grugel, B. Breit, Org. Lett. 20 (2018) 1066-1069. DOI:10.1021/acs.orglett.7b04035 |
| [54] |
N. Gommermann, P. Knochel, Chem. Commun. 2004 (2004) 2324-2325. |
| [55] |
Q. Liu, H. Xu, Y. Li, et al., Nat. Commun. 12 (2021) 19-28. DOI:10.53388/mdm2021112421 |
| [56] |
Z.B. Li, T.D. Liu, L. Pu, J. Org. Chem. 72 (2007) 4340-4343. DOI:10.1021/jo070091j |
| [57] |
W. Chen, J.H. Tay, J. Ying, X.Q. Yu, L. Pu, J. Org. Chem. 78 (2013) 2256-2265. DOI:10.1021/jo3026065 |
| [58] |
W.C. Huang, W. Liu, X.D. Wu, J. Ying, L. Pu, J. Org. Chem. 80 (2015) 11480-11484. DOI:10.1021/acs.joc.5b02185 |
| [59] |
Y.M. Li, Y.Q. Tang, X.P. Hui, L.N. Huang, P.F. Xu, Tetrahedron 65 (2009) 3611-3614. DOI:10.1016/j.tet.2009.03.005 |