 2023, Vol. 34
2023, Vol. 34 


b Research Institute of Applied Chemistry, Jiangxi Academy of Sciences, Nanchang 330096, China
Syngas, a mixture of CO and H2, is widely used as the reactant to produce a variety of organic chemicals, which depends on the used catalysts. Fundamental understanding of catalytic CO hydrogenation reactions is of great importance, but meanwhile, has remained as a long-standing challenge due to the ultrahigh complexity. Theoretical calculations have demonstrated great capacity in fundamentally studying complex catalytic CO hydrogenation reactions [1, 2].
Cu-based catalysts have been widely used to catalyze CO hydrogenation reactions either to methanol or to methane, and Cu/ZnO/Al2O3 catalyst is the commercial catalyst for CO hydrogenation to methanol [3, 4]. This has inspired extensive fundamental investigations of CO hydrogenation reactions on Cu-based surfaces, but arguments still exist whether the Cu-ZnO interface or the CuZn alloy is the active site for catalyzing CO hydrogenation to methanol reaction [5-9]. Recently, both experimental and theoretical calculation studies showed the in-situ formed CuZn alloys in Cu/ZnO/Al2O3 catalysts under CO hydrogenation reaction conditions as the active structure, whose formation is preferred at stepped Cu sites and whose catalytic performance is sensitive to the Cu structure [10-12]. However, comprehensive studies of elementary surface reaction networks of CO hydrogenation reactions to both methanol and methane on stepped Cu and CuZn alloy surfaces have been few. Cu(211) and Cu(611) stepped facets are very common on the low-indexed Cu(111) and Cu(100) surfaces, respectively. In this Letter, we report a comparative DFT calculation study of elementary surface reaction networks of CO hydrogenation on stepped Cu(211), Cu(611), ZnCu(211) and ZnCu(611) surfaces. The results demonstrate surface structure-sensitive catalytic activity and reaction mechanisms.
DFT calculations were carried out using the Vienna ab-initio simulation package (VASP) [13, 14] using the projector-augment wave (PAW) method [15]. The nonlocal exchange correlation energy was performed via the generalized gradient approximation (GGA) and PBE functional [16, 17]. 11-layer slab with (1 × 4) surface cell and 22-layer slab with (1 × 2) surface cell were performed to simulate stepped surface Cu(211) and Cu(611). A (1 × 4)-11-layer Cu(211) slab and a (1 × 2)-22-layer Cu(611) slab with 1/4 Cu substituted by Zn on the step edge, denoted as ZnCu(211) and ZnCu(611), were used to simulate CuZn alloy [18]. The adsorption energies (Eads) was calculated at the most stable configuration as Eads = Eads/sub – Eads – Esub, in which Eads/sub, Eads and Esub are the energy of the optimized adsorption system of adsorbate and substrate, adsorbate in the gas phase, and the clean substrate, respectively. All transition states (TSs) were located by the force reversed method [19] and climbing-image nudged elastic band method (CINEB) [20, 21]. A value of U − J = 4.7 eV was used for the correction of the on-site Coulomb repulsion of 3d electrons of Zn atoms [22-24].
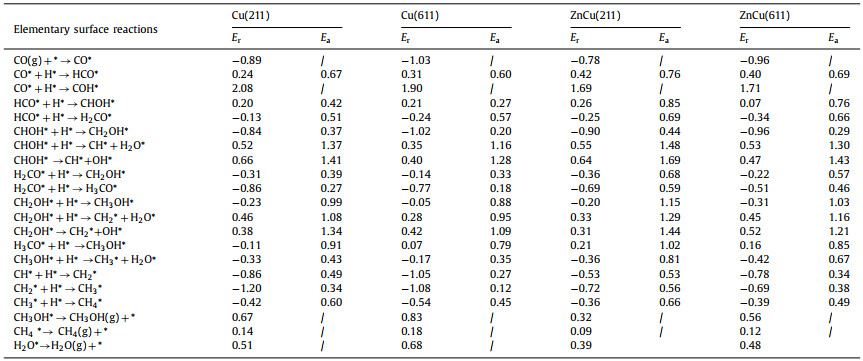
Fig. 1 illustrates calculated elementary surface reaction network of CO hydrogenation reactions on Cu and CuZn alloy surfaces during our theoretical calculations. Direct CO dissociation on Cu surfaces was reported to exhibit large barriers [25, 26], thus only H-assisted CO activation paths were considered, including CO* + H* → HCO* and CO* + H* → COH*. The elementary surface reaction network consists of competing elementary surface reactions between HnCO* + H* → Hn+1CO* and HnCO* + H* → CHnOH* (n = 0, 1, 2), competing elementary surface reactions among CHnOH* → CHn* + OH*, CHnOH* + H* → CHn* + H2O* (n = 1, 2, 3) and CHnOH* + H* → CHn+1OH* (n = 1, 2), elementary surface reactions of CHn* + H* → CHn+1* (n = 1, 2, 3), adsorption of CO and H2, and desorption of CH3OH*, CH4* and H2O*.

|
Download:
|
| Fig. 1. Calculated elementary surface reaction network of CO hydrogenation reactions on Cu and CuZn alloy surfaces. The red arrows represent the elementary surface reactions of CHnOH* + H* → CHn* + H2O* (n = 1, 2, 3). | |
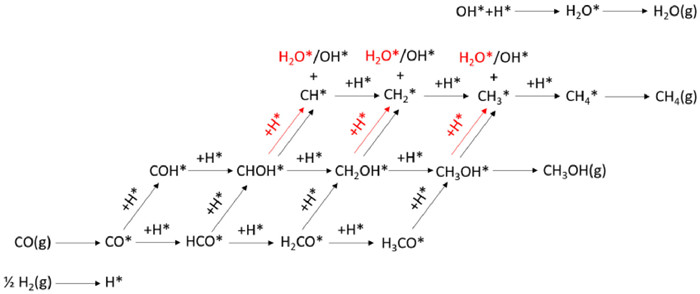
Fig. 2 presents the optimized structures of stepped Cu(211), Cu(611), ZnCu(211) and ZnCu(611) surfaces. Adsorptions of surface intermediates included in Fig. 1 on Cu and ZnCu surfaces were calculated. The optimized adsorption configurations are shown in Fig. S1 (Supporting information) and the corresponding adsorption energies are summarized in Table S1 (Supporting information). Cu(611) and ZnCu(611) bind surface intermediates more strongly than Cu(211) and ZnCu(211), respectively, consistent with the fact that a more open surface with more coordination-unsaturated surface atoms is more reactive. Meanwhile, Cu(211) and Cu(611) bind surface intermediates more strongly than ZnCu(211) and ZnCu(611), respectively, suggesting that alloying with Zn weakens the reactivity of Cu surface. CH4 adsorption energies on Cu(211), Cu(611), ZnCu(211) and ZnCu(611) surfaces are all within −0.2 eV, however, CH3OH adsorption energy decreases from −0.67 eV on Cu(211) to −0.32 eV on ZnCu(211) and from −0.83 eV on Cu(611) to −0.56 eV on ZnCu(611), and H2O adsorption energy decreases from −0.51 eV on Cu(211) to −0.39 eV on ZnCu(211) and from −0.68 eV on Cu(611) to −0.48 eV on ZnCu(611). Thus, desorption of produced CH3OH* and H2O* species on ZnCu alloy surfaces is much easier than on corresponding Cu surfaces.

|
Download:
|
| Fig. 2. Side and top views of optimized structures of stepped Cu(211), Cu(611), ZnCu(211) and ZnCu(611) surfaces. | |
Activation energy (Ea) and reaction energy (Er) of elementary surface reactions shown in Fig. 1 were also calculated. The calculated Ea and Er are summarized in Table 1 and the optimized configurations of involved transition states are shown in Figs. S2–S4 (Supporting information). The Ea and Er of H2 dissociation were calculated to be 0.11 and −0.46 eV on Cu(211), 0.06 and −0.57 eV on Cu(611), 0.12 and −0.35 eV on ZnCu(211), and 0.08 and −0.44 eV on ZnCu(611), respectively. Therefore, H2 dissociation is facilitated both kinetically and thermodynamically on a more open Cu surface. Meanwhile, alloying with Zn slightly increases the barrier and decreases the reaction energy of H2 dissociation on the Cu surface.
|
|
Table 1 Calculated activation energies (Ea in eV) and reaction energies (Er in eV) of elementary surface reactions involved in CO hydrogenation on Cu(211), Cu(611), ZnCu(211) and ZnCu (611) surfaces. |
{kind=link}
{kind=link}
The Er of CO* + H* → HCO* and CO* + H* → COH* reactions were calculated as 0.24 and 2.08 eV on Cu(211), 0.31 and 1.90 eV on Cu(611), 0.42 and 1.69 eV on ZnCu(211), and 0.40 and 1.71 eV on ZnCu(611), respectively. Meanwhile, the Ea of CO* + H* → HCO* reaction were calculated as 0.67, 0.60, 0.76 and 0.69 eV on Cu(211), Cu(611), ZnCu(211) and ZnCu(611), respectively. This indicates that the Ea of CO* + H* → COH* reaction on Cu and ZnCu surfaces is much larger than that of CO* + H* → HCO* and those of other elementary surface reactions. Thus, the CO* + H* → COH* reaction does not likely occur under CO hydrogenation reactions over Cu and ZnCu alloy surfaces.
The Er/Ea of HCO* + H* → CHOH* and HCO* + H* → H2CO* are 0.20/0.42 eV and −0.13/0.51 eV on Cu(211) and 0.21/0.27 eV and −0.24/0.57 eV on Cu(611), respectively. Thus HCO* + H* → CHOH* is endothermic on Cu surfaces whereas HCO* + H* → H2CO* is exothermic; however, HCO* + H* → CHOH* exhibits a smaller barrier than HCO* + H* → H2CO*, the difference of which is 0.09 eV on Cu(211) and 0.30 eV on Cu(611). The Er/Ea of H2CO* + H* → CH2OH* and H2CO* + H* → H3CO* are −0.31/0.39 eV and −0.86/0.27 eV on Cu(211) and −0.14/0.33 eV and −0.77/0.18 eV on Cu(611), respectively, suggesting that H2CO* + H* → H3CO* is preferred both thermodynamically and kinetically over H2CO* + H* → CH2OH*. The Er/Ea of H3CO* + H* → CH3OH* are −0.11/0.91 eV on Cu(211) and 0.07/0.79 eV on Cu(611).
The Er/Ea of CHOH* + H* → CH2OH*, CHOH* + H* → CH* + H2O* and CHOH* → CH*+OH* are −0.84/0.37 eV, 0.52/1.37 eV and 0.66/1.41 eV on Cu(211), and −1.02/0.20 eV, 0.35/1.16 eV and 0.40/1.28 eV on Cu(611), respectively. Meanwhile, the Er/Ea of CH2OH* + H* → CH3OH*, CH2OH* + H* → CH2* + H2O* and CH2OH* → CH2*+OH* are −0.23/0.99 eV, 0.46/1.08 eV and 0.38/1.34 eV on Cu(211), and −0.05/0.88 eV, 0.28/0.95 eV and 0.42/1.09 eV on Cu(611), respectively. Thus, CHnOH* + H* → CHn+1OH* (n = 1, 2) is preferred both thermodynamically and kinetically over CHnOH* + H* → CHn* + H2O* and CHnOH* → CHn* + OH*, particularly for the CHOH* species.
The Er/Ea of CH3OH* + H* → CH3* + H2O* are −0.33/0.43 eV on Cu(211) and −0.17/0.35 eV on Cu(611), respectively, whereas the Er of CH3OH* desorption is 0.67 and 0.83 eV on Cu(211) and Cu(611), respectively. Thus the CH3OH* prefers both thermodynamically and kinetically the reaction of CH3OH* + H* → CH3* + H2O* to produce CH3* over the desorption to produce CH3OH. The Er/Ea of CH3* + H* → CH4* are −0.42/0.60 eV on Cu(211) and −0.54/0.45 eV on Cu(611), respectively, and the Er of CH4* desorption is 0.14 and 0.18 eV on Cu(211) and Cu(611), respectively.
The Er/Ea of HCO* + H* → CHOH* and HCO* + H* → H2CO* are 0.26/0.85 eV and −0.25/0.69 eV on ZnCu(211) and 0.07/0.76 eV and −0.34/0.66 eV on ZnCu(611), respectively. The Er/Ea of H2CO* + H* → CH2OH* and H2CO* + H* → H3CO* are −0.36/0.68 eV and −0.69/0.59 eV on ZnCu(211) and −0.22/0.57 eV and −0.51/0.46 eV on ZnCu(611), respectively. Thus, HnCO* + H* → Hn+1CO* is preferred both thermodynamically and kinetically over HnCO* + H* → CHnOH* (n = 1, 2). The Er/Ea of H3CO* + H* → CH3OH* are 0.21/1.02 eV on ZnCu(211) and 0.16/0.85 eV on ZnCu(611).
The Er/Ea of CHOH* + H* → CH2OH*, CHOH* + H* → CH* + H2O* and CHOH* → CH*+OH* are −0.90/0.44 eV, 0.55/1.48 eV and 0.64/1.69 eV on ZnCu(211), and −0.96/0.29 eV, 0.53/1.30 eV and 0.47/1.43 eV on ZnCu(611), respectively. Meanwhile, the Er/Ea of CH2OH* + H* → CH3OH*, CH2OH* + H* → CH2* + H2O* and CH2OH* → CH2*+OH* are −0.20/1.15 eV, 0.33/1.29 eV and 0.31/1.44 eV on ZnCu(211), and −0.31/1.03 eV, 0.45/1.16 eV and 0.52/1.21 eV on ZnCu(611), respectively. Thus, CHnOH* + H* → CHn+1OH* (n = 1, 2) is preferred both thermodynamically and kinetically over CHnOH* + H* → CHn* + H2O* and CHnOH* → CHn* + OH*, particularly for the CHOH* species.
The Er/Ea of CH3OH* + H* → CH3* + H2O* are −0.36/0.81 eV on ZnCu(211) and −0.42/0.67 eV on ZnCu(611), respectively, whereas the Er of CH3OH* desorption is 0.32 and 0.56 eV on ZnCu(211) and ZnCu(611), respectively. Thus the CH3OH* prefers both thermodynamically and kinetically the desorption to produce CH3OH over the reaction of CH3OH* + H* → CH3* + H2O* to produce CH3*.
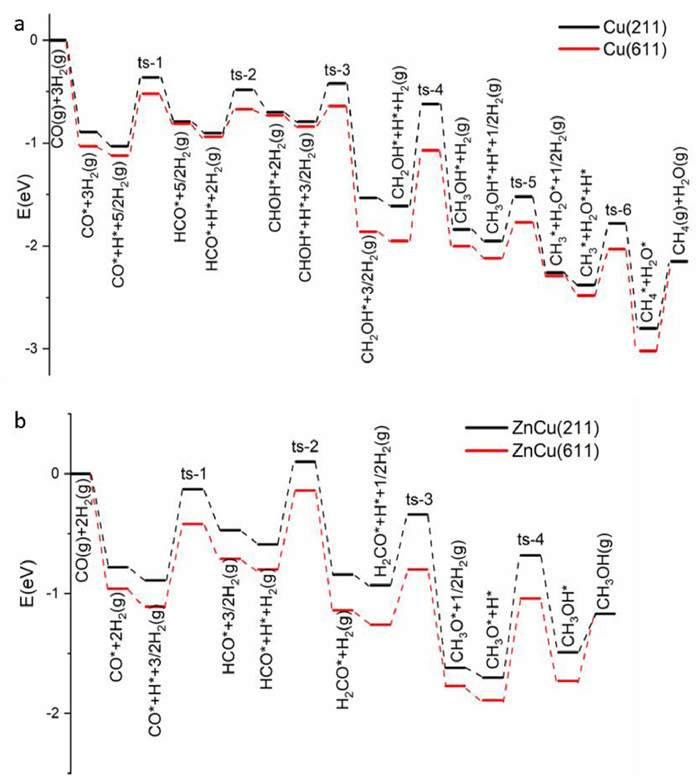
Based on the above calculation results, the energetic favorable reaction path of CO hydrogenation reaction follows CO* → HCO* → CHOH* → CH2OH* → CH3OH* → CH3* → CH4* → CH4 on Cu(211) and Cu(611) surfaces (Fig. 3a). The CH2OH* + H* → CH3OH* is the rate-limiting step, and its barrier is smaller on Cu(611) (0.88 eV) than on Cu(211) (0.99 eV). Meanwhile, the barriers of CH3OH* + H* → CH3* + H2O* and CH3OH* desorption determine the catalytic selectivity of CH3OH and CH4, being 0.43 and 0.67 eV on Cu(211) and 0.35 and 0.83 eV on Cu(611), respectively, thus Cu(611) surface should exhibit a higher CH4 selectivity than Cu(211) surface. Therefore, Cu(611) surface is more active and selective than Cu(211) surface in catalyzing CO hydrogenation reaction to CH4. However, it is noteworthy that the reaction path of HCO* → H2CO* → H3CO* → CH3OH* → CH3* → CH4* → CH4 likely occurs on Cu(211) due to the not much differences between HCO* + H* → CHOH* and HCO* + H* → H2CO*, but not on Cu(611).

|
Download:
|
| Fig. 3. Energetic favorable reaction path of CO hydrogenation reaction on (a) Cu(211) and Cu(611) surfaces and (b) ZnCu(211) and ZnCu(611) surfaces. | |
{kind=link}
Alloying Cu surface with Zn leads to a larger barrier of HCO* + H* → CHOH* than of HCO* + H* → H2CO* and a larger barrier of CH3OH* + H* → CH3* + H2O* than CH3OH* desorption, thus the energetic favorable reaction path of CO hydrogenation reaction follows CO* → HCO* → H2CO* → H3CO* → CH3OH* → CH3OH on ZnCu(211) and ZnCu(611) surfaces (Fig. 3b). The H3CO* + H* → CH3OH* reaction is the rate-limiting step, and its barrier is smaller on ZnCu(611) (0.85 eV) than on ZnCu(211) (1.02 eV). However, the barriers of CH3OH* + H* → CH3* + H2O* and CH3OH* desorption are 0.81 and 0.32 eV on ZnCu(211) and 0.67 and 0.56 eV on ZnCu(611), respectively. Therefore, ZnCu(611) surface is more active but less selective than ZnCu(211) surface in catalyzing CO hydrogenation reaction to CH3OH.
In summary, via a comparative DFT calculation study of elementary surface reaction network, we demonstrate the energetic favorable reaction path and structure-sensitivity of CO hydrogenation reactions on stepped Cu(211), Cu(611), ZnCu(211) and ZnCu(611) surfaces. CO hydrogenation reaction on Cu(211) and Cu(611) surfaces favorably produces CH4 following the reaction path of CO* → HCO* → CHOH* → CH2OH* → CH3OH* → CH3* → CH4* → CH4. Cu(611) surface is more active and selective than Cu(211) surface. Alloying Cu surface with Zn leads to a larger barrier of HCO* + H* → CHOH* than of HCO* + H* → H2CO* and a larger barrier of CH3OH* + H* → CH3* + H2O* than CH3OH* desorption, resulting in the favorite reaction path of CO hydrogenation reaction on ZnCu(211) and ZnCu(611) surfaces as CO* → HCO* → H2CO* → H3CO* → CH3OH* → CH3OH to produce CH3OH. ZnCu(611) surface is more active but less selective than ZnCu(211) surface.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 91745202, 92145302), the Chinese Academy of Sciences, the Changjiang Scholars Program of Ministry of Education of China.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107809.
| [1] |
W. Chen, R. Pestman, B. Zijlstra, I.A.W. Filot, E.J.M. Hensen, ACS Catal. 7 (2017) 8050-8060. DOI:10.1021/acscatal.7b02757 |
| [2] |
W. Chen, I.A.W. Filot, R. Pestman, E.J.M. Hensen, ACS Catal. 7 (2017) 8061-8071. DOI:10.1021/acscatal.7b02758 |
| [3] |
K.C. Waugh, Catal. Today 15 (1992) 51-75. DOI:10.1016/0920-5861(92)80122-4 |
| [4] |
K. Klier, Adv. Catal. 31 (1982) 243-313. DOI:10.1080/02786820290038546 |
| [5] |
S. Kuld, et al., Science 352 (2016) 969-974. DOI:10.1126/science.aaf0718 |
| [6] |
F. Studt, et al., ChemCatChem 7 (2015) 1105-1111. DOI:10.1002/cctc.201500123 |
| [7] |
Y. Choi, K. Futagami, T. Fujitani, J. Nakamura, Catal. Lett. 73 (2001) 27-31. DOI:10.1023/a:1009074219286 |
| [8] |
D. Laudenschleger, H. Ruland, M. Muhler, Nat. Commun. 11 (2020) 3898. DOI:10.1038/s41467-020-17631-5 |
| [9] |
N.D. Nielsen, J. Thrane, A.D. Jensen, J.M. Christensen, Catal. Lett. 150 (2020) 1427-1433. DOI:10.1007/s10562-019-03036-7 |
| [10] |
M. Behrens, et al., Science 336 (2012) 893-897. DOI:10.1126/science.1219831 |
| [11] |
Z. Zhang, X. Chen, J. Kang, et al., Nat. Commun. 12 (2021) 4331. DOI:10.1038/s41467-021-24621-8 |
| [12] |
Y.F. Shi, P.L. Kang, C. Shang, Z.P. Liu, J. Am. Chem. Soc. 144 (2022) 13401-13414. DOI:10.1021/jacs.2c06044 |
| [13] |
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758-1775. |
| [14] |
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [15] |
P.E. Blochl, Phys. Rev. B 50 (1994) 17953-17979. DOI:10.1103/PhysRevB.50.17953 |
| [16] |
J.P. Perdew, Y. Wang, Phys. Rev. B 45 (1992) 13244-13249. DOI:10.1103/PhysRevB.45.13244 |
| [17] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [18] |
F. Studt, F. Abild-Pedersen, J.B. Varley, J.K. Nørskov, Catal. Lett. 143 (2013) 71-73. DOI:10.1007/s10562-012-0947-5 |
| [19] |
K.J. Sun, Y.H. Zhao, H.Y. Su, W.X. Li, Theor. Chem. Acc. 131 (2012) 1118-1127. DOI:10.1007/s00214-012-1118-x |
| [20] |
G. Henkelman, H. Jonsson, J. Chem. Phys. 113 (2000) 9978-9985. DOI:10.1063/1.1323224 |
| [21] |
G. Henkelman, B.P. Uberuaga, H. Jonsson, J. Chem. Phys. 113 (2000) 9901-9904. DOI:10.1063/1.1329672 |
| [22] |
A. Janotti, C.G. Van de Walle, Phys. Rev. B 76 (2007) 165202-165223. DOI:10.1103/PhysRevB.76.165202 |
| [23] |
F. Liao, X.P. Wu, J. Zheng, et al., Catal. Sci. Technol. 6 (2016) 7698-7702. DOI:10.1039/C6CY01832G |
| [24] |
F. Liao, X.P. Wu, J. Zheng, et al., Green Chem. 19 (2017) 270-280. DOI:10.1039/C6GC02366E |
| [25] |
X.H. Zhao, Y.W. Li, J.G. Wang, C.F. Huo, J. Fuel Chem. Technol. 39 (2011) 956-960. DOI:10.1016/S1872-5813(12)60004-8 |
| [26] |
G. Jia, L. Ling, R. Zhang, B. Wang, Mol. Catal. 518 (2022) 112071. DOI:10.1016/j.mcat.2021.112071 |