 2023, Vol. 34
2023, Vol. 34 


The selective oxidation of sulfides to sulfoxides, as one of the most commercially valuable chemical reactions, has recently attracted abounding attention from industry and academia, not only because such transformation can purify toxic and harmful sulfur-containing chemicals, but also because the sulfoxide products can be used as important intermediates for the synthesis of high value-added fine chemicals [1-4]. In order to replace the longstanding industrial synthesis techniques that directly use stoichiometric hypervalent chromium and manganese reagents, periodic acid and its salts, and other hazardous reagents as oxidants, many sustainable and green catalytic systems have been extensively developed for the sulfide-sulfoxide transformation thus far [2-4]. For instance, various simple metal catalysts (MOFs, inorganic oxides, etc.) and metal-free catalysts (COFs, ionic liquids, etc.) have been successfully synthesized and served as catalysts for molecular oxygen-, hydrogen peroxide-, or tert-butyl hydroperoxide-based oxidation of sulfides in mild conditions [4-7]. Regrettably, common disadvantages of most of above systems are insufficiently high sulfoxide selectivity and catalytic efficiency.
Over the past decades, the charming multifunctional nano-metal-oxygen clusters (also known as polyoxometalates, POMs) mostly composed of early transition metals in high oxidation states have been intensively demonstrated to be a class of high-efficiency catalysts that exhibit excellent catalytic activity in many fields [8-11]. Extensive works have shown that POM-based homogeneous catalytic systems are feasible for the selective oxidation of sulfides to corresponding sulfoxides; however, the poor recyclability hindered their further application [12-15]. Although some subsequent studies reveal that the loading strategies can alleviate the problems of catalysts recovery and products separation to a certain extent, it is often accompanied by new obstacles such as low loading efficiency and leaching of risk POMs [16]. In addition, the ill-defined structure and inhomogeneous catalytic sites of these supported catalytic systems also bring certain limitations to the exploration of their catalytic mechanisms.
Research interests of late years have gradually focused on the construction of versatile porous materials [17-19]. Among them, POM-based metal-organic frameworks (POMOFs), as a burgeoning class of crystalline porous material, are highly sought after since their unique structures and properties, as well as their potential applications in various fields [20-22]. Many excellent works have shown that POMOFs exhibit high catalytic activity and excellent recycling performance in catalyzing liquid-phase reactions such as alcohol oxidation, phenol oxidation, olefin oxidation, water splitting and CO2 reduction [22-27]. With aspect of sulfide-sulfoxide transformation using H2O2 or TBHP as oxidant, a few polyoxovanadate/polyoxomolybdate-based MOFs, such as (MMo6)(NH2)2(4-tts) (M = Mn, Fe, Co), Cu(2-mim)4V2O6, Co(dtba)V2O6, Co2(4-ra)0.5V4O12·3DMF·5H2O, have been successfully proved to be effective catalysts [28-32]. Contrastively, in our uninterrupted literature and structure retrieval, we found that polyoxotungstate-based MOFs have hardly been reported in selective oxidation of sulfides. Furthermore, the oxidant efficiencies of most POMOFs catalytic systems reported so far for sulfide oxidation are relatively low. Therefore, the development of efficient heterogeneous catalysts is highly desirable.
In this work, we chose α-Keggin-type phosphotungstates as the inorganic building blocks, copper(Ⅱ) ions as the metal node of MOFs, and 2-(3-pyridinyl)-1H-imidazole-4,5-dicarboxylic acid (H3pidc) as multi-dentate bridging ligands, with or without the participation of vanadium-based mineralizers, successfully isolated two pseudo-polymorphic porous POM-pillared MOFs, H3n[Cu3(pidc)2(H2O)2.5]2[PW12O40]n·xH2O (n = 1.5, x = 6 for 1, n = 1, x = 12 for 2). As expected, both 1 and 2 exhibited excellent catalytic activity in the TBHP-based oxidation of various sulfide substrates. Particularly, in the catalytic oxidation of methylphenyl sulfide (MPS), the yield of corresponding sulfoxide can be close to 100% within 30–50 min. The catalytic activity measured by the turnover frequencies (TOF) are as high as 600–1000 h−1. The mechanism analysis reveal that the catalytic process may involve a "POM-Cu-microenvironment" ternary synergistic effect.
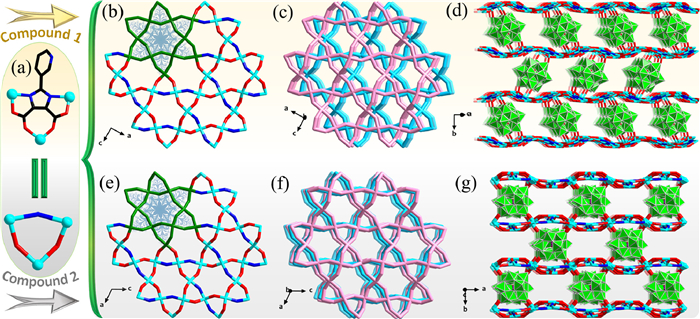
Single crystal X-ray diffraction analyses reveal that both 1 and 2 were crystallized in the monoclinic system with space group P21/c and C2/c, respectively (Table S1 in Supporting information). In 1, there are six crystallographically independent Cu(Ⅱ) ions, which can be classified into two categories from the geometric configuration, namely Cu1, Cu3 and Cu6, with a pentacoordinate trigonal bipyramidal hexahedral geometry, as well as Cu2, Cu4 and Cu5, with a hexacoordinate tetragonal bipyramidal octahedral geometry (Fig. S7 in Supporting information). One can note that all four crystallographically independent H3pidc ligands adopt the "4,5-imidazoledicarboxylic acid" mode, coordinating to three Cu2+ ions arranged in a triangle (denoted as {Cu3pidc} subunit, Cu⋯Cu: 5.55–6.28 Å) (Fig. 1a). Interestingly, six {Cu3pidc} subunits are interconnected to form a blooming petal-like 12-nuclear metal-organic unit, which is further extended into a fascinating and slightly undulating two-dimensional (2D) metal-organic layer through shared {Cu3pidc} subunits (Fig. 1b). As shown in Fig. 1c, these 2D layers can be stacked on each other along the b-axis with a 2.5 Å displacement between adjacent layers and a layer spacing of 11.9 Å (Fig. S8 in Supporting information). Furthermore, the adjacent 2D layers are eventually connected by tetradentate PW12 anions (denoted as μ4-PW12) to form a porous POM-pillared metal-organic framework (Fig. 1d). The channel window size of three-dimensional (3D) framework is 5.5 × 9.5 Å, which is significantly larger than the molecular size of the MPS (5.3 × 6.2 Å) (Table S3 in Supporting information). In addition, the other crystallographically independent bidentate PW12 polyanion (denoted as μ2-PW12) in 1 hang on both sides of the 2D metal-organic layer; however, μ2-PW12 does not participate in the construction of 3D framework (Fig. S9 in Supporting information).

|
Download:
|
| Fig. 1. Structures of 1 and 2: (a) Coordination mode of ligands. (b, e) Two-dimensional metal organic layers. (c, f) The stacked two-dimensional metal organic layers. (d, g) Three-dimensional porous POM-pillared MOFs. Color code: {WO6} octahedron, green; Cu, turquoise; O, red; N, blue; C, black; simplified ligand, red and blue; simplified 2D layer, rose or sky blue. | |
{kind=link}
For 2, the three copper ions have four crystallographic characteristics, that is, Cu1, Cu2, Cu3, and Cu4 with 100%, 50%, 50% and 100% occupancies, respectively (Fig. S11 in Supporting information). Like 1, the two crystallographically independent H3pidc ligand in compound 2 also present the coordination form of "4,5-imidazole dicarboxylic acid", and also form a similar {Cu3pidc} subunit with three copper ions (Cu⋯Cu: 5.54–6.19 Å) (Fig. 1a). Also, a petal-like dodecaucleated metal-organic unit and a more regular slightly undulating 2D layer similar to those in 1 can also be obtained from the {Cu3pidc} subunit (Fig. 1e). Likewise, such 2D layers can also be stacked "face-to-face" along the b-axis (the interlayer spacing of about 10.5 Å) (Fig. S12 in Supporting information); however, a significant difference from 1 is that there is no displacement bias between adjacent 2D layers (Fig. 1f). Finally, POMs also severed as tetradentate inorganic ligands to provide four μt-O atoms (two on each side) to connect adjacent metal-organic layers to form porous POM-pillared MOFs (4.5 × 9.0 Å) (Fig. 1g). Unlike the two μt-O atoms on the same side in 1 from two adjacent W atoms, the two μt-O atoms on the same side in 2 are located on two non-adjacent W atoms (Fig. S13 in Supporting information).
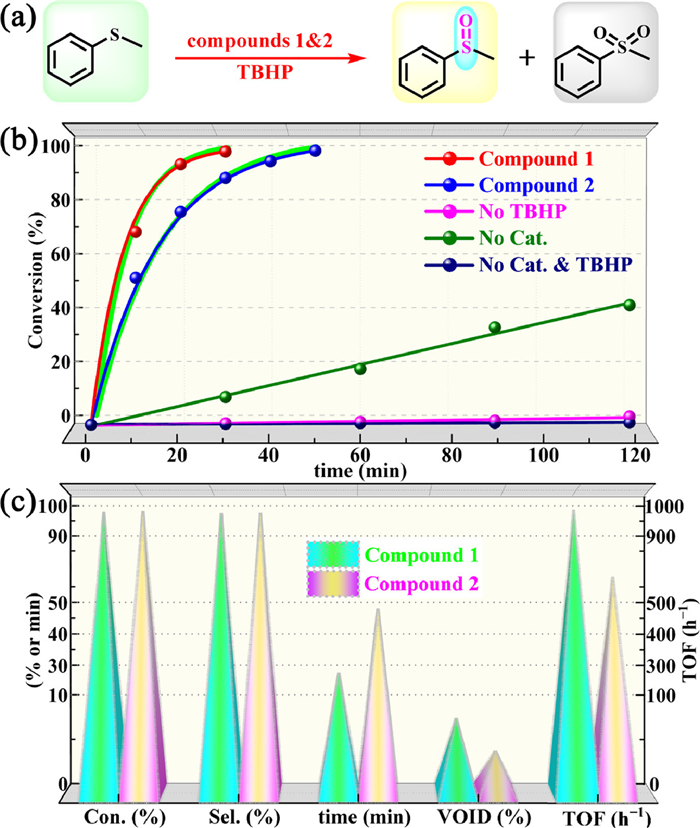
In the present study, we substantiated the 1 and 2 have vast potential for application in TBHP-based oxidation of sulfides to sulfoxides (Fig. 2a). One can see from Fig. 2b that 1 can convert > 99% of MPS within 30 min, accompanied by 98.9% selectivity of methylphenylene sulfoxide (MPSO) (standard reaction conditions: nsub.: ncat.: nTBHP = 1:0.2%: 1.4, reacted at 50 ℃ in 0.5 mL of MeCN). The blank experiment without oxidant showed almost no conversion of the substrate even if the reaction time was prolonged. Although good conversion of the substrate can be achieved for a sufficiently long time using only oxidizing agents, the selectivity for sulfoxide is mediocre (Table S4 in Supporting information, entry 2). These results indicated that both 1 and TBHP are indispensable for the efficient conversion of sulfides to sulfoxides.

|
Download:
|
| Fig. 2. (a) Schematic diagram of oxidation of MPS catalyzed by compounds 1 and 2. (b) Control experiments of under standard conditions. (c) Comparison of catalytic performances of compounds 1 and 2 (Selectivity to MPSO, and void space was calculated by the PLATON program). | |
{kind=link}
Compound 2 can also achieve nearly 100% MPSO yield under standard conditions, but it takes 50 min to complete the transformation (Fig. 2b). Moreover, the catalytic activity of 2 expressed in TOF is 600 h-1, which is significantly lower than that of 1 (TOF = 1000 h-1) (Fig. 2c). Thanks to their well-defined structures and well-ordered components, we can easily dissect the reasons leading to their different activities from the perspective of structure. As can be seen from the above structural description, 1 and 2 have highly similar metal-organic layers, but their 3D structures have different porosities due to the different coordination modes of PW12 polyanions. Calculations with the PLATON program revealed that 1 contains 8.3% void-space (10,815.0 Å3 per unit cell) accessible by solvent molecules, which is nearly twice that of 2 (4.7%, 9315.7 Å3 per unit cell) (Fig. 2c). Higher porosity can theoretically offer more active sites and more efficient mass transfer efficiency. Importantly, 1 also showed more excellent catalytic activities in contrast to the vast majority POMOFs, even most POM-based catalysts reported so far. (Tables S5 and S6 in Supporting information) [33,34]. This work also provides guidance for designing more efficient heterogeneous catalysts.
The difference in the activity of 1 and 2 was also further investigated by kinetic analyses (Figs. S14 and S15 in Supporting information). One can see that the corresponding curves were all well fitted to the first-order kinetic characteristics. Based on Arrhenius equation, the activation energy (Ea) of MPS oxidation to MPSO catalyzed by 1 was 30.2 kJ/mol at 40–70 ℃, which was significantly less than that of compound 2 (Ea = 43.9 kJ/mol), and the relationship between them was also consistent with the above catalytic results. Out of these data, the corresponding activation entropies (ΔS≠) can also be further calculated as −202.3 J mol-1 K-1 for 1, as well −163.5 J mol-1 K-1 for 2 by the Eyring equation (Table S7 in Supporting information). The negative entropies of 1 and 2 may be due to the loss of degrees of freedom of the system caused by the adsorption coordination of substrates during the reaction. Moreover, the more negative the value was, the stronger the corresponding coordination ability was in theory, which further confirmed the more prominent catalytic activity of 1 [35,36].
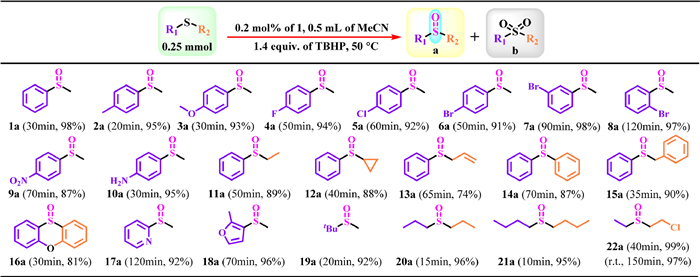
Motivated by the above excellent results, a series of differently functionalized sulfides were selected as substrates to explore the general applicability of the title catalysts under optimum reaction conditions (Scheme 1). One can notice that 1 demonstrated excellent catalytic activity among a wide range of substituted-phenyl methyl sulfides (Scheme 1, 1a-10a). The satisfactory sulfoxide yields (almost all > 90%) can be obtained within sufficient reaction time regardless of electron-withdrawing or an electron-donating groups-functionalized substrates. And for substituted-phenyl methyl sulfides, the position of the substituent group has a certain effect on the reactivity of the substrates, specifically speaking, the reactivity from large to small in the order of para-position > meta-position > ortho-position (Scheme 1, 6a-8a). In addition, 1 also showed good tolerance to phenyl alkyl sulfides (Scheme 1, 11a-15a). For N- and O-heterocyclic groups functionalized sulfoxides, excellent yields (81%–96%) can still be obtained (Scheme 1, 16a-18a). Aliphatic sulfides show higher reactivity than aromatic sulfides, with typical examples such as tert-butylmethyl sulfide, dipropyl sulfide, and n-butylsulfide harvesting > 90% of the target product in 10–20 min (Scheme 1, 19a-21a). In particular, 2-chloroethyl ethyl sulfide (CEES) is often studied as a simulant of sulfur mustard that is one of the most vicious chemical warfare agents. Herein, we first studied the catalytic degradation of CEES catalyzed by 1 at room temperature (r.t.); however, the catalytic results were not very ideal (Scheme 1, 22a). A relatively considerable catalytic result required to be obtained at higher reaction temperatures, and CEES can achieve nearly 100% conversion to nontoxic 2-chloroethyl ethyl sulfoxide (CEESO) within 40 min (Scheme 1, 22a). In addition, 1 also exhibited potential catalytic oxidative desulfurization performance, and the catalytic results showed that benzothiophene, dibenzothiophene, and 4,6-dimethyl-dibenzothiophene can be rapidly converted into sulfones and sulfoxides (Table S8 in Supporting information).

|
Download:
|
| Scheme 1. Oxidation of different sulfides to the corresponding sulfoxides catalyzed by 1. | |
{kind=link}
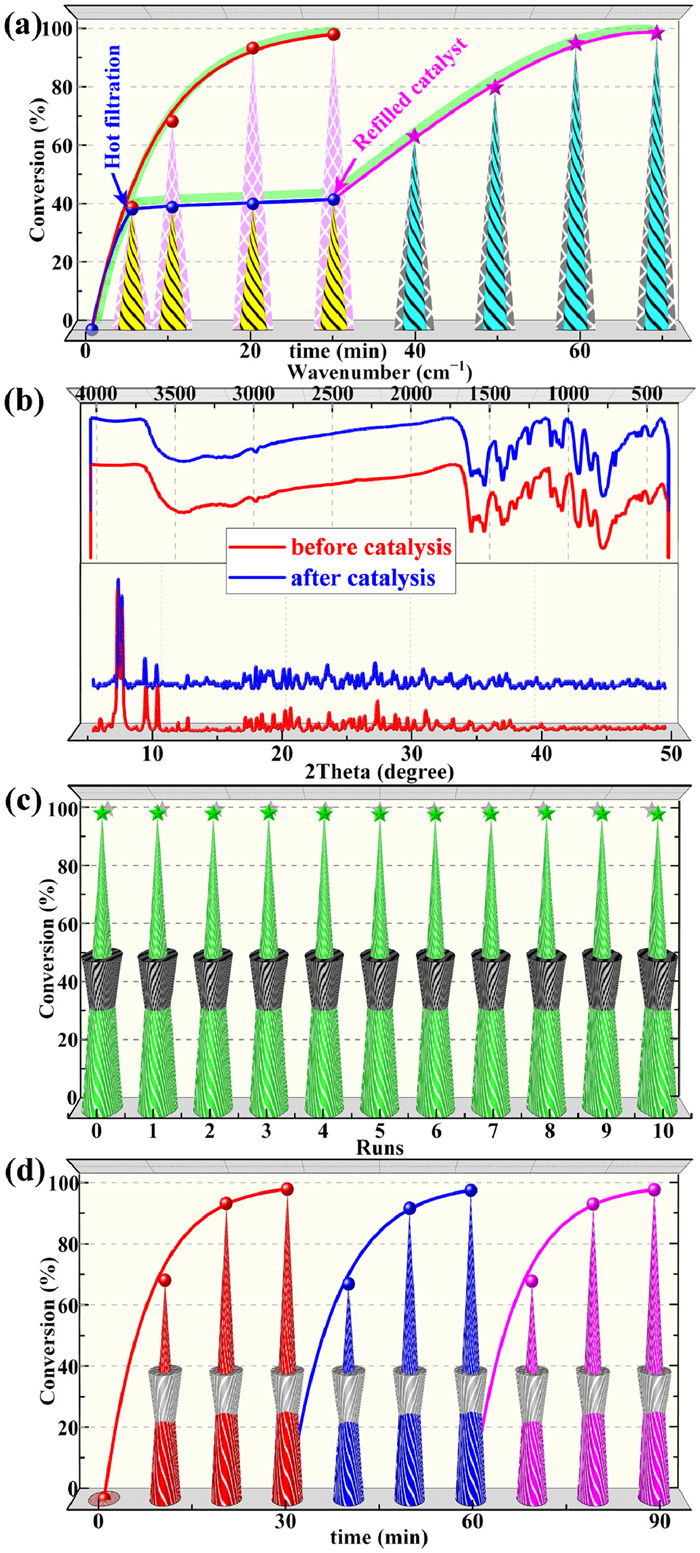
In order to better prove its heterogeneous nature, the catalysts in the system were quickly removed when the reaction proceeded for 5 min, and the remaining filtrate continued to react in the standard solution for another 25 min. The results showed only a negligible increase in conversion (< 5%), probably due to the sole oxidation of the oxidant (Fig. 3a). Moreover, the transformation was nearly complete again within 30 min when the catalyst was refilled (Fig. 3a). The UV–vis spectrum of the filtrate soaked in 1 under standard catalytic conditions did not detect the characteristic peaks of POMs and Cu ions, which also further demonstrated the heterogeneous nature of the 1 (Fig. S16 in Supporting information). As Fig. 3b showed, the catalysts recovered by simple filtration has highly consistent FT-IR spectra and PXRD patterns with the newly synthesized catalysts, demonstrating the excellent structural stability of the title catalysts. Cycling experiments show that the catalytic activity can still be maintained at the level of fresh catalysts in at least 10 cycles (Fig. 3c). The continuous cycling experiments also corroborated the similar stability conclusion that the conversion of MPS hardly decreases after three consecutive experiments (Fig. 3d). Additionally, the almost constant conversion after three cycles in the kinetic regime also showed the reliable reusability of 1 (Fig. S18 in Supporting information). Taken together, these results were good confirmation of the stability and heterogeneity of the title catalysts.

|
Download:
|
| Fig. 3. Stability of 1: (a) Hot filtration experiment for MPS oxidation. (b) FT-IR spectra and PXRD patterns before and after the catalytic reaction. (c) Recycle experiment of MPS oxidation. (d) Continuous cycle experiments. | |
{kind=link}
Comparative experiments show that the superior activities of 1 and 2 may originate from the synergistic effect of the POMs site and the Cu(Ⅱ) site, while assisted by the structural micro-environment (Table S9 in Supporting information). Many well-known POMs without other transition metal substitutions are usually metal atoms on the skeleton as active sites, and the real active species are the metal-peroxides in the TBHP- and H2O2-based oxidation reactions [37]. To verify the formation of active metal-peroxide, UV–vis spectroscopic characterization is considered to be a simple and reliable method. The specific performance is as follows: after the DMSO solution of 1 is treated with a few drops of oxidant, the absorption peak corresponding to the O→W charge transfer transition in the ultraviolet region undergoes an obvious red-shift phenomenon (Fig. S19a in Supporting information). In addition, copper ions are prone to Fenton-like reactions and generate reactive hydroxyl radicals in the presence of hydrogen peroxide or TBHP [17]. In the free radical trapping experiments, we noticed that the addition of butylated hydroxytoluene and isopropanol inhibited the reaction to some extent, while the addition of diphenylamine and 2,2,6,6-tetramethylpiperidin-1-yl)oxyl hardly stalled the reaction (Fig. S19b in Supporting information). To further demonstrate the formation of hydroxyl radicals, the photoluminescence technology was employed. As expected, the fluorescence of terephthalic acid was switched on after the introduction of TBHP and 1, indicating that 1 can indeed activate TBHP to generate hydroxyl radicals (Fig. S20 in Supporting information). These results suggested that a copper-based radical pathway may also exist in the catalytic system. Based on these analyses, a possible dual-path mechanism emerges as the times required (Scheme 2). For the pathway A, the polyoxoanion was first activated by the oxidant to form the active peroxy species (A1). Immediately, the nucleophilic sulfur atom in the substrates attacked A1 to generate an unsteady intermediate (A2), which could easily transform into another unstable intermediate A3 through intramolecular electron migration. Finally, A3 could quickly release the corresponding sulfoxide. During this process, the polyoxoanions returned to its original form, completing the catalytic cycle. And for pathway B, the Cu(Ⅱ) site first interacted with the substrates and was reduced to Cu(Ⅰ) while generating the electron-deficient intermediate (B1). Subsequently, TBHP (tBuOOH) can easily gain an electron from Cu(Ⅰ) to generate tert-butoxide anion (tBuO-) and active hydroxyl radical (•OH), and Cu(Ⅰ) was simultaneously oxidized to the initial Cu(Ⅱ) state. The •OH rapidly reacted with B1 to form B2, which can be removed a hydrogen proton to obtain the final sulfoxide products. And the protons are further reacted with tBuO- to generate the tert-butanol by-products.

|
Download:
|
| Scheme 2. Proposed TBHP-based oxidation processes of sulfides to sulfoxides catalyzed by title catalysts. | |
{kind=link}
Briefly, we established a mineralizer-regulated self-assembly strategy and successfully synthesized two pseudo-polymorphic porous POM-pillared MOFs in this paper. Catalytic studies indicated that 1 with higher porosity exhibits more prominent catalytic activity for TBHP-based oxidation of sulfides to sulfoxides than 2. The structure-activity relationship showed that the larger porosity of 1 can led to more active sites and more efficient mass transfer process. Kinetic analysis also showed that 1 had stronger interaction with the substrates and lower activation energy in the catalytic MPS oxidation reaction. Moreover, the catalytic activities and structural stabilities of both catalysts did not change significantly over at least 10 cycles. The excellent catalytic activity of title catalysts originated from the efficient synergistic catalysis of POM sites and Cu sites assisted by the structural microenvironment. This work not only set a precedent for the use of polyoxotungstate-based MOFs for the selective oxidation of sulfides, but also provided convincing guidance for the design of more efficient heterogeneous catalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21371027, 20901013), Natural Science Foundation of Liaoning Province (No. 2015020232), and Fundamental Research Funds for the Central Universities (Nos. DUT19LK01, DUT15LN18).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107856.
| [1] |
N. Wang, P. Saidhareddy, X. Jiang, Nat. Prod. Rep. 37 (2020) 246-275. DOI:10.1039/C8NP00093J |
| [2] |
S. Otocka, M. Kwiatkowska, L. Madalinska, P. Kielbasinski, Chem. Rev. 117 (2017) 4147-4181. DOI:10.1021/acs.chemrev.6b00517 |
| [3] |
M. Ja'fari, S.L. Ebrahimi, M.R. Khosravi-Nikou, Ultrason. Sonochem. 40 (2018) 955-968. DOI:10.1016/j.ultsonch.2017.09.002 |
| [4] |
N.S. Bobbitt, M.L. Mendonca, A.J. Howarth, et al., Chem. Soc. Rev. 46 (2017) 3357-3385. DOI:10.1039/C7CS00108H |
| [5] |
Y. Meng, Y. Luo, J.L. Shi, et al., Angew. Chem. Int. Ed. 59 (2020) 3624-3629. DOI:10.1002/anie.201913091 |
| [6] |
X. Lang, J. Zhao, X. Chen, Angew. Chem. Int. Ed. 55 (2016) 4697-4700. DOI:10.1002/anie.201600405 |
| [7] |
S. Hou, N. Chen, P. Zhang, S. Dai, Green Chem. 21 (2019) 1455-1460. DOI:10.1039/C8GC03772H |
| [8] |
N. Li, J. Liu, B.X. Dong, Y.Q. Lan, Angew. Chem. Int. Ed. 59 (2020) 20779-20793. DOI:10.1002/anie.202008054 |
| [9] |
J.C. Liu, J.-.F. Wang, Q. Han, et al., Angew. Chem. Int. Ed. 60 (2021) 11153-11157. DOI:10.1002/anie.202017318 |
| [10] |
W. An, X. Zhang, J. Niu, et al., Chin. Chem. Lett. 33 (2022) 4400-4404. DOI:10.1016/j.cclet.2021.12.021 |
| [11] |
Y. Ma, F. Gao, W. Xiao, et al., Chin. Chem. Lett. 33 (2022) 4395-4399. DOI:10.1016/j.cclet.2021.12.023 |
| [12] |
C. Yang, Q. Jin, H. Zhang, et al., Green Chem. 11 (2009) 1401-1405. DOI:10.1039/b912521n |
| [13] |
J. Dong, J.F. Hu, Y.N. Chi, et al., Angew. Chem. Int. Ed. 56 (2017) 4473-4477. DOI:10.1002/anie.201700159 |
| [14] |
X. Wang, K. Brunson, H. Xie, et al., J. Am. Chem. Soc. 143 (2021) 21056-21065. DOI:10.1021/jacs.1c11208 |
| [15] |
Y. Yang, F. Tao, L. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2625-2629. DOI:10.1016/j.cclet.2021.09.093 |
| [16] |
J. Li, Z. Yang, S. Li, et al., J. Ind. Eng. Chem. 82 (2020) 1-16. DOI:10.1016/j.jiec.2019.10.020 |
| [17] |
S. Chang, Y. Chen, H. An, et al., Green Chem. 23 (2021) 8591-8603. DOI:10.1039/D1GC03061B |
| [18] |
C. Zou, Z. Zhang, X. Xu, et al., J. Am. Chem. Soc. 134 (2012) 87-90. DOI:10.1021/ja209196t |
| [19] |
H. Furukawa, K.E. Cordova, M. O'Keeffe, O.M. Yaghi, Science 341 (2013) 1230444. DOI:10.1126/science.1230444 |
| [20] |
Q. An, Z. Xu, W. Shang, et al., ACS Appl. Bio Mater. 5 (2022) 1222-1229. DOI:10.1021/acsabm.1c01252 |
| [21] |
Y.R. Huang, X.L. Lin, B. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 16911-16916. DOI:10.1002/anie.202104333 |
| [22] |
D.Y. Du, J.S. Qin, S.L. Li, et al., Chem. Soc. Rev. 43 (2014) 4615-4632. DOI:10.1039/C3CS60404G |
| [23] |
S. Chang, H. An, Y. Chen, et al., ACS Appl. Mater. Interfaces 11 (2019) 37908-37919. DOI:10.1021/acsami.9b14928 |
| [24] |
H. Tian, S. Liu, Z. Zhang, et al., ACS Sustain. Chem. Eng. 9 (2021) 4660-4667. DOI:10.1021/acssuschemeng.1c00389 |
| [25] |
D. Li, P. Ma, J. Niu, J. Wang, Coord. Chem. Rev. 392 (2019) 49-80. DOI:10.1016/j.ccr.2019.04.008 |
| [26] |
Y. Song, Y. Peng, S. Yao, et al., Chin. Chem. Lett. 33 (2022) 1047-1050. DOI:10.1016/j.cclet.2021.08.045 |
| [27] |
K. Yang, Y.Y. Hu, L.Y. Li, et al., Nano Energy 74 (2020) 104851. DOI:10.1016/j.nanoen.2020.104851 |
| [28] |
J. Li, X. Huang, S. Yang, et al., Cryst. Growth Des. 15 (2015) 1907-1914. DOI:10.1021/acs.cgd.5b00086 |
| [29] |
X. Wang, T. Zhang, Y.H. Li, et al., Inorg. Chem. 59 (2020) 17583-17590. DOI:10.1021/acs.inorgchem.0c02798 |
| [30] |
Y. Chen, S. Chang, H. An, et al., ACS Sustain. Chem. Eng. 9 (2021) 15683-15693. DOI:10.1021/acssuschemeng.1c06433 |
| [31] |
B.B. Lu, J. Yang, Y.Y. Liu, J.F. Ma, Inorg. Chem. 59 (2017) 11710-11720. |
| [32] |
R. Ma, N. Liu, T.T. Lin, et al., J. Mater. Chem. A 8 (2020) 8548-8553. DOI:10.1039/D0TA02443K |
| [33] |
H.Y. An, Y.J. Hou, L. Wang, et al., Inorg. Chem. 56 (2017) 11619-11632. DOI:10.1021/acs.inorgchem.7b01564 |
| [34] |
X.L. Hao, Y.Y. Ma, H.Y. Zang, et al., Chem. Eur. J. 21 (2015) 3778-3784. DOI:10.1002/chem.201405825 |
| [35] |
H.G.T. Ly, G. Fu, A. Kondinski, et al., J. Am. Chem. Soc. 140 (2018) 6325-6335. DOI:10.1021/jacs.8b01902 |
| [36] |
D. Wang, J. Jiang, M.-.Y. Cao, et al., Nano Res. 15 (2021) 3628-3637. |
| [37] |
S. Chang, Y. Chen, H. An, et al., ACS Appl. Mater. Interfaces 13 (2021) 21261-21271. DOI:10.1021/acsami.1c02558 |