 2023, Vol. 34
2023, Vol. 34 


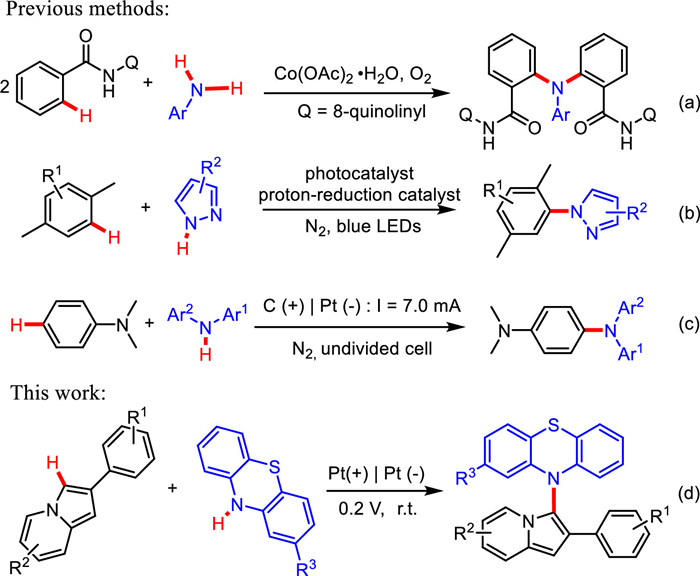
Triarylamines were becoming increasingly important due to their hole-transporting capabilities and unusual stable radical cations [1–3]. Among them, N-aryl phenothiazine derivatives played an important role in organic light-emitting diodes [4–7], sensitized solar cells [8–11] and photochromic materials [12,13]. The methods for efficient synthesis of N-aryl phenothiazines via the formation of C−N bonds were fascinating. Over the past decade, strategies for constructing C−N bonds have been intensively developed [14–19]. Du et al. reported an efficient protocol for C−H amination of arenes with anilines to synthesize triarylamines catalyzed by Co(OAc)2·4H2O (Scheme 1a) [18]. A method for photo-induced C−H/N−H cross-coupling between arenes and azoles without oxidant was proposed by Niu and co-workers [19]. The C−N bonds were constructed conveniently under synergistic effect of photocatalyst and proton-reduction catalyst at room temperature (Scheme 1b). However, part of classical synthetic methods inevitably required chemical oxidants, transition metal catalysts or photosensitizers, which were not in line with the characteristics of sustainability and green chemistry.

|
Download:
|
| Scheme 1. Construction of the C–N bonds. | |
As an environmentally friendly and efficient synthetic method, electrochemistry provided new possibilities for green organic synthesis [20–25]. In recent years, electrocatalytic C−H functionalization to construct C−N bonds has been successfully established [26–31]. In 2019, Lei and co-workers developed an elegant strategy for C–H/N–H cross-coupling with hydrogen evolution to synthesize triarylamine derivatives from electron-rich aromatic hydrocarbons and diarylamines (Scheme 1c) [32].
Encouraged by these exciting results and with our continuation of research on electrochemical synthesis [33–35], we envisioned that phenothiazines and 2-phenylindolizines might be promising to synthesize novel compounds for optoelectronic materials, due to both structural motifs of the compounds had excellent electrochemical and photochemical characteristics [36–38]. Herein, we reported an efficient and convenient method for electrochemical oxidative C−H/N−H cross-coupling of 2-phenylindolines with phenothiazines to synthesize a series of novel N-aryl phenothiazine derivatives (Scheme 1d). Redox properties of the reactions were investigated by cyclic voltammetry and in situ FTIR [39–41].
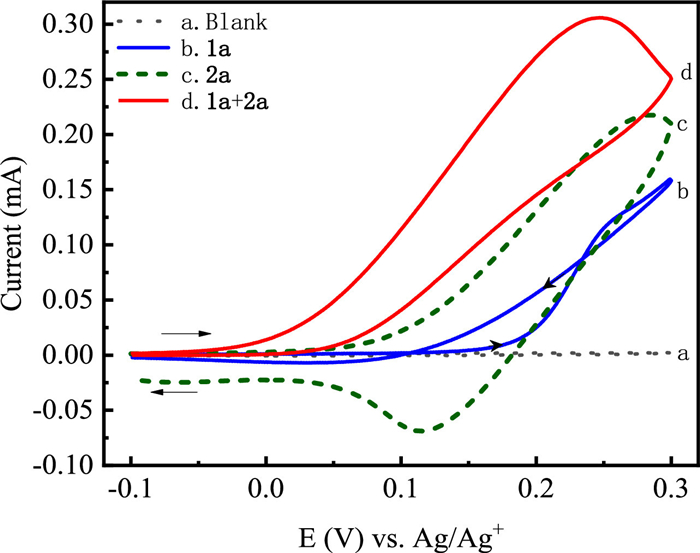
Cyclic voltammetry experiments were carried out to study the redox behavior of the reactants. As shown in Fig. 1, no distinct electrochemical reaction peaks existed in the cyclic voltammogram of the LiClO4/DMF solution (blank solution). With the addition of 2-phenylindolizine (1a) into the blank solution, the oxidation current began to increase from about 0.15 V and the oxidation peak was clearly observed at about 0.25 V. A classic nucleation loop exhibited near 0.20 V on the backward scanning and the surface of the anode was covered by a yellow thin film during the cyclic voltammetry experiment. It was related to the homogeneous formation of oligomeric products from 1a [36,42]. When phenothiazine (2a) was added into the blank solution alone, 2a began to be oxidized at about 0.03 V. An obvious oxidation peak and a reduction peak appeared at 0.29 V and 0.11 V, respectively. The oxidation of 2a was quasi-reversible and just one electron was transferred in the oxidation process [43,44]. As shown in Fig. 1d, with the addition of 1a and 2a in the blank solution, the onset oxidation potential shifted negatively to −0.05 V and a new oxidation peak with higher current was noticed at 0.23 V. Most importantly, the loop at about 0.2 V for the oxidation of 1a disappeared and thin film was not formed on the anode surface, which indicated that the polymerization of 1a was inhibited. Meanwhile, the reduction peak of 2a was not observed. The above phenomena suggested that 1a reacted with 2a during the process of cyclic voltammetry experiment.

|
Download:
|
| Fig. 1. Cyclic voltammograms recorded in 0.1 mol/L LiClO4/DMF (15 mL) from −0.1 V to 0.3 V at 50 mV/s scan rate with (a) blank solution; (b) 1a (0.016 mol/L); (c) 2a (0.013 mol/L); (d) 1a (0.016 mol/L) and 2a (0.013 mol/L). | |
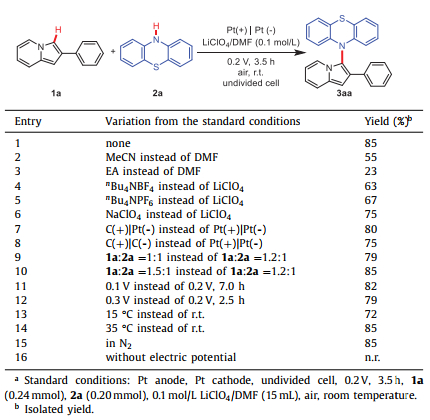
As an initial attempt, the cross-coupling of 1a with 2a was selected as the model reaction to screen out the optimal conditions and the results were summarized in Table 1. When 1a (0.24 mmol) reacted with 2a (0.20 mmol) in 0.1 mol/L LiClO4/DMF solution at the potential of 0.2 V for 3.5 h, the yield of 10-(2-phenylindolizin-3-yl)−10H-phenothiazine (3aa) could reach 85% (entry 1). The X-ray crystallographic coordinates for structure of 3aa were shown in Fig. S1 (Supporting information). Replacing DMF with acetonitrile and ethyl acetate, the yields were decreased to 55% and 23%, respectively. It might be related to the poor solubility of 1a in acetonitrile and the weak conductivity of ethyl acetate solution (entries 2 and 3). The supporting electrolytes had a certain influence on the reaction. Three other supporting electrolytes (nBu4NBF4, nBu4NPF6, NaClO4) were tested instead of LiClO4 in the standard conditions, the yield of 3aa was decreased slightly (entries 4–6). When a graphite rod was employed as the anode instead of a platinum plate, 80% yield of 3aa could be obtained (entry 7). Using graphite rods both as the anode and cathode was less effective than platinum plates (entry 8). When the dosage of 1a was reduced to 1.0 equiv., the yield of 3aa was decreased to 79% (entry 9). While 1.5 equiv. of 1a could not improve the yield of 3aa obviously (entry 10). Though the reactions were also carried out well at 0.1 V or 0.3 V, no better yields were achieved (entries 11 and 12). When the reaction temperatures were 15 ℃ and 35 ℃, the yields of 3aa were 72% and 85%, respectively. Therefore, room temperature was suitable for the reaction (entries 13 and 14). In addition, nitrogen atmosphere had little effect on the reaction (entry15), and almost no reaction took place without electric potential (entry 16).
|
|
Table 1 Optimization of the reaction conditions. |
{kind=link}
{kind=link}
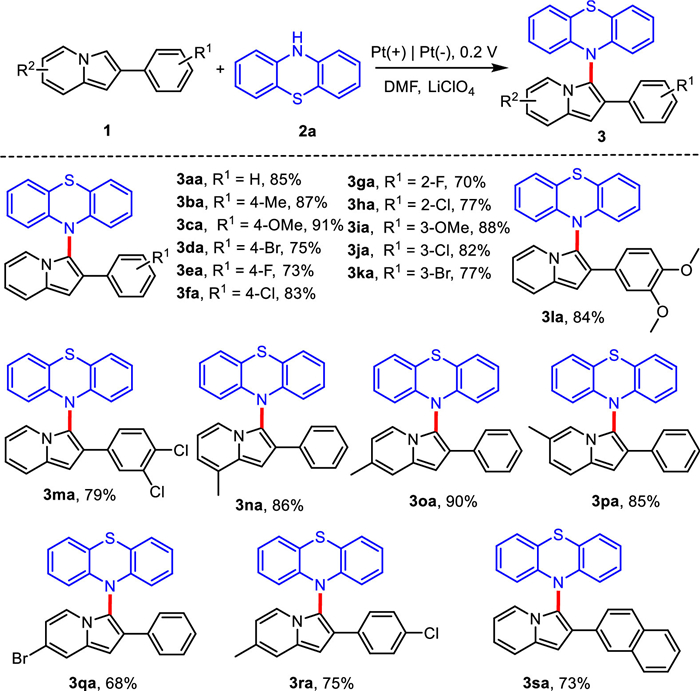
With the optimized conditions in hand, we caried out an exploration of the substrate range. A series of 2-phenylindolazines were investigated and the results were summarized in Scheme 2. C2-substituted phenyl moiety of 2-phenylindolizines bearing –Me or –OMe at para-position reacted with 1a to generate corresponding products in excellent yields (3ba and 3ca). The presence of –Cl, –Br, –F groups at para-position of C2-substituted phenyl moiety slightly reduced the synthetic efficiency, and the desired coupling products 3da-3fa were isolated in 75%−83% yields. Meanwhile, the different position of substituent group had little effect on the reaction, which was shown in the yields of 3ga-3ka and 3na-3qa. The 2-phenylindolazines with disubstituent on phenyl moiety also performed well to generate the corresponding products in 84% and 79%, respectively (3la and 3ma). In addition, the 2-(4-chlorophenyl)−7-methylindolizine and 2-(naphthalen-2-yl)indolizine could afford good results under optimized conditions (3ra and 3sa). Either electron-withdrawing groups or electron-donating groups substituted on 1a, the coupling reactions could be carried out smoothly.

|
Download:
|
| Scheme 2. Substrate scope of 2-phenylindolizines. Reaction conditions: 1 (0.24 mmol), 2a (0.20 mmol), Pt(+)|Pt(-), 0.1 mol/L LiClO4/DMF (15 mL), an undivided cell, 0.2 V, r.t., 3.5 h. | |
{kind=link}
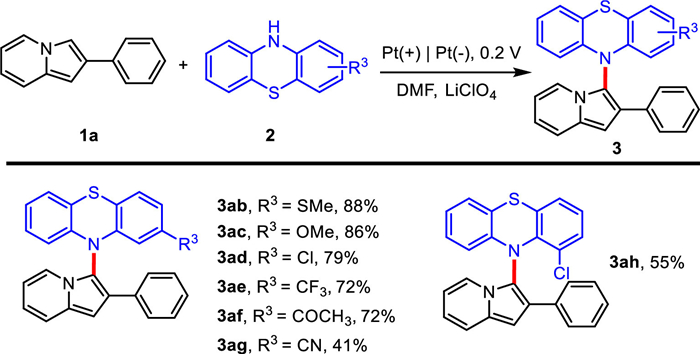
Subsequently, the coupling of a variety of phenothiazines (2b-2g) with 1a was investigated, and the results were shown in Scheme 3. 2-Methylthio-10 H-phenothiazine (2b) and 2-methoxy-10 H-phenothiazine (2c) were suitable for the reaction and the desired products 3ab and 3ac could be isolated in 86% and 88% yields, respectively. When phenothiazines bearing –Cl, –CF3, –COCH3 substituents at the 2-position were submitted to the cross-coupling, the yields of 3ad-3af were higher than 72%. However, 10 H-phenothiazine-2-carbonitrile (2g) could not achieve a satisfactory result, the yield of 3ag was only 41%. When –Cl was substituted at 1-position of phenothiazine, the corresponding product 3ah was obtained in 55% yield. It indicated the steric hindrance has some influence on the reaction.

|
Download:
|
| Scheme 3. Substrate scope of phenothiazines. Reaction conditions: 1a (0.24 mmol), 2 (0.20 mmol), Pt(+)|Pt(-), 0.1 mol/L LiClO4/DMF (15 mL), an undivided cell, 0.2 V, r.t., 3.5 h. | |
{kind=link}
In general, 2-phenylindolazines or phenothiazines with electron-donating groups showed higher activity than that with electron-withdrawing groups. In addition, the cross-coupling of 2-phenylindolizines with phenothiazines under galvanostatic electrolysis was also investigated. However, the potential tended to increase during galvanostatic electrolysis continuously, and high potential might cause electro-polymerization of 2-phenylindolazines. Therefore, the yields of the products with galvanostatic electrolysis were lower than that with potentiostatic electrolysis. The results of galvanostatic electrolysis were described in the Supporting Information. In addition, the constant cell potential electrolysis with two electrodes was also investigated. When the cell potentials were 3.0 V, 4.0 V and 5.0 V, the yields of 3aa were 12%, 65% and 49%, respectively.
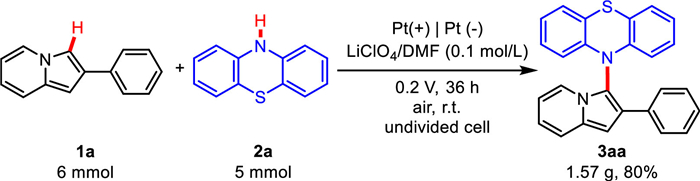
To evaluate the scalability of this electrocatalytic system, the electrochemical cross-coupling of 1a with 2a was performed on 5 mmol scale. The reaction was carried out at 0.2 V for 36 h and the desired product 3aa was obtained in 80% yield (Scheme 4). It showed that this electrochemical method was feasible in practical synthesis.

|
Download:
|
| Scheme 4. Gram-scale synthesis. | |
{kind=link}
In situ FTIR spectroscopy was an important tool to study the reaction characteristics of electrode surface and helped us to analyze the consumption of reactants and the formation of products. The cross-coupling reaction of 1a (0.72 mmol) with 2a (0.60 mmol) was investigated by in situ FTIR technique, which was implemented in 0.1 mol/L LiClO4/DMF solution at potentials varied from −300 mV to 500 mV. As shown in Fig. 2, three important positive bands at 1574, 1474 and 1260 cm−1 and four negative bands at 1562, 1547, 1285 and 1198 cm−1 could be observed. The positive-going band at 1574 cm−1 was attributed to N−H bending vibration of 2a which was very characteristic [45], and the bond at 1260 cm−1 was related to C−N stretching vibration of 2a [46]. It confirmed that 2a was consumed during the electrochemical oxidation process. Moreover, the positive-going band located at 1474 cm−1 could be attributed to a characteristic of the nitrogen heterocycle skeleton vibration [47], which showed the participation of 1a in this reaction.

|
Download:
|
| Fig. 2. In situ FTIR spectra collected during the reaction of 1a (0.72 mmol) and 2a (0.60 mmol) in 15 mL 0.1 mol/L LiClO4/DMF at potentials varied from −300 mV to 500 mV. | |
{kind=link}
It was noteworthy that due to the conjugation effect of heterocyclic aromatic hydrocarbon and lone pair electrons of nitrogen, the skeleton vibration of aromatic ring would red-shift [48]. Two negative-going bonds at 1562 and 1547 cm−1 were related to aromatic skeleton vibrations of 3aa. The negative-going bond at 1285 cm−1 was attributed to the C−N stretching vibration of 3aa. In addition, the band located at 1198 cm−1 was assigned to ClO4 − of the supporting electrolyte [33,49]. Therefore, 3aa could be synthesized from 1a and 2a successfully at low potential.
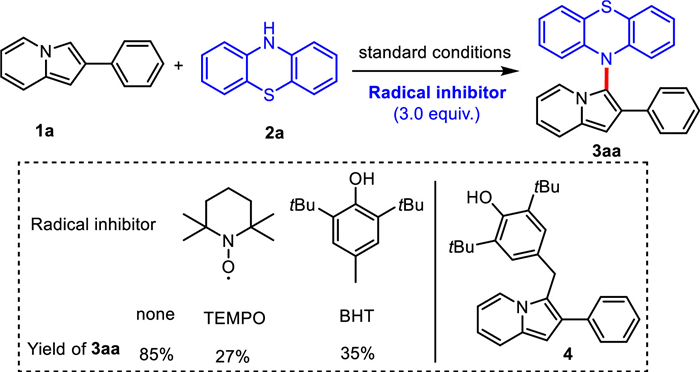
In order to gain insight into the process of dehydrogenative C–H/N–H cross-coupling between 1a and 2a, a few radical-trapping control experiments were conducted as shown in Scheme 5. Three equivalents of the radical scavenger 2,2,6,6-tetramethylpiperidine-oxyl (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) was added to the standard conditions and formation of 3aa was inhibited. It showed that a radical pathway might be involved in the coupling reaction of 2-phenylindolizines with phenothiazines during the electrochemical oxidation process. Furthermore, a BHT-trapped complex 4 was detected by electrospray ionization mass spectrometry (ESI-MS) analysis (see Supporting information for details).

|
Download:
|
| Scheme 5. Control experiments. | |
{kind=link}
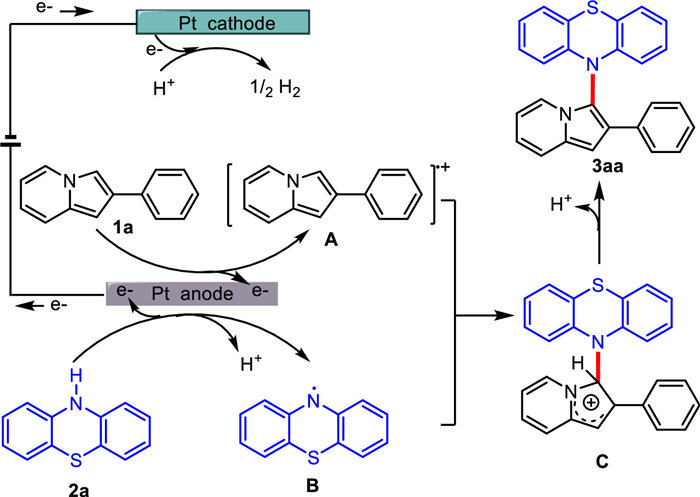
Based on the results of the above experiments and literatures [50–53], a plausible mechanism for this reaction was revealed in Scheme 6. Initially, the anodic oxidation of 1a occurred to form the 2-phenylindolizine cationic radical A, and 2a was oxidized at platinum plate anode to form phenothiazine radical B. Then, cationic radical A coupled with radical B to form intermediate C and C−N bond was constructed. The intermediate C underwent deprotonation to generate 3aa. At the same time, the protons were reduced at the cathode to generate H2.

|
Download:
|
| Scheme 6. Proposed reaction mechanism. | |
{kind=link}
In summary, we have developed a novel and atom-economical method for the synthesis of N-aryl phenothiazine derivatives via electrochemical oxidative C–H/N–H cross-coupling of 2-phenylindolizines with phenothiazines under undivided electrolytic conditions. The cross-coupling reactions were carried out smoothly without external oxidants and catalysts. The C–N bonds were constructed at a low potential which could reduce the side reactions such as electrochemical polymerization of the 2-phenylindolizines. In addition, the results of cyclic voltammetry, in situ FTIR and control experiments were used to further understand the oxidation process in the reactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the generous financial support of the National Natural Science Foundations of China (Nos. 22178321, 21773211 and 21776260).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107935.
| [1] |
J. Wang, K. Liu, L. Ma, X. Zhan, Chem. Rev. 116 (2016) 14675-14725. DOI:10.1021/acs.chemrev.6b00432 |
| [2] |
C.S. Wu, Y. Chen, J. Mater. Chem. 20 (2010) 7700-7709. DOI:10.1039/c0jm00707b |
| [3] |
K. Zhang, H.J. Niu, C. Wang, et al., J. Electroanal. Chem. 682 (2012) 101-109. DOI:10.1016/j.jelechem.2012.06.018 |
| [4] |
B. Wang, Y. Zheng, T. Wang, D.G. Ma, Q. Wang, Org. Electron. 88 (2021) 106012. DOI:10.1016/j.orgel.2020.106012 |
| [5] |
G. Bagdžiūnas, G. Grybauskaitė, N. Kostiv, et al., RSC Adv. 6 (2016) 61544-61554. DOI:10.1039/C6RA12692H |
| [6] |
I. Marghad, F. Bencheikh, C. Wang, et al., RSC Adv. 9 (2019) 4336-4343. DOI:10.1039/C8RA10393C |
| [7] |
B. Wang, X. Qiao, Z. Yang, et al., Org. Electron. 59 (2018) 32-38. DOI:10.1016/j.orgel.2018.04.045 |
| [8] |
C.J. Yang, Y.J. Chang, M. Watanabe, Y.S. Hon, T.J. Chow, J. Mater. Chem. 22 (2012) 4040-4049. DOI:10.1039/c2jm13961h |
| [9] |
A.F. Buene, D.M. Almenningen, J. Mater. Chem. 9 (2021) 11974-11994. |
| [10] |
D. Devadiga, M. Selvakumar, P. Shetty, et al., Int. J. Energy Res. 45 (2021) 6584-6643. DOI:10.1002/er.6348 |
| [11] |
E. Kavery, S. Muruganantham, S. Prabhu, et al., Optik 208 (2020) 164046. DOI:10.1016/j.ijleo.2019.164046 |
| [12] |
C. Qian, Z. Ma, J. Liu, et al., Chem. Asian. J. 16 (2021) 3713-3718. DOI:10.1002/asia.202101002 |
| [13] |
B. Ramakrishna, K. Narayanaswamy, S.P. Singh, P.R. Bangal, Asian J. Org. Chem. 7 (2018) 2254-2262. DOI:10.1002/ajoc.201800338 |
| [14] |
C.M. Chan, Y.C. Chow, W.Y. Yu, Synth. Stuttgart 52 (2020) 2899-2921. |
| [15] |
D.G. Ma, S. Zhai, Y. Wang, A.A. Liu, C.C. Chen, Front. Chem. 7 (2019) 635. DOI:10.3389/fchem.2019.00635 |
| [16] |
Y. Wu, J.A. Chen, Q. Li, W.T. Wei, Chin. J. Org. Chem. 40 (2020) 589-597. DOI:10.6023/cjoc201909032 |
| [17] |
N. Zheng, H.X. Zheng, T. Li, W.T. Wei, ChemSusChem 14 (2021) 5340-5358. DOI:10.1002/cssc.202102243 |
| [18] |
C. Du, P.X. Li, X.J. Zhu, et al., ACS Catal. 7 (2017) 2810-2814. DOI:10.1021/acscatal.7b00262 |
| [19] |
L.B. Niu, H. Yi, S.C. Wang, et al., Nat. Commun. 8 (2017) 14226. DOI:10.1038/ncomms14226 |
| [20] |
B. Elsler, D. Schollmeyer, K.M. Dyballa, R. Franke, S.R. Waldvogel, Angew. Chem. Int. Ed. 53 (2014) 5210-5213. DOI:10.1002/anie.201400627 |
| [21] |
T. Morofuji, A. Shimizu, J. Yoshida, J. Am. Chem. Soc. 135 (2013) 5000-5003. DOI:10.1021/ja402083e |
| [22] |
J. Yoshida, K. Kataoka, R. Horcajada, A. Nagaki, Chem. Rev. 108 (2008) 2265-2299. DOI:10.1021/cr0680843 |
| [23] |
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492-2521. DOI:10.1039/c3cs60464k |
| [24] |
Y. Yu, Y. Yuan, H. Liu, et al., Chem. Commun. 55 (2019) 1809-1812. DOI:10.1039/C8CC09899A |
| [25] |
J.W. Wu, Y. Zhou, Y.C. Zhou, C.W. Chiang, A.W. Lei, ACS Catal. 7 (2017) 8320-8323. DOI:10.1021/acscatal.7b03551 |
| [26] |
S. Tang, S. Wang, Y. Liu, H. Cong, A.W. Lei, Angew. Chem. Int. Ed. 57 (2018) 4737-4741. DOI:10.1002/anie.201800240 |
| [27] |
Y.C. Wu, S.S. Jiang, R.J. Song, J.H. Li, Chem. Commun. 55 (2019) 4371-4374. DOI:10.1039/C9CC01332F |
| [28] |
S. Zhang, L.J. Li, M.Y. Xue, et al., Org. Lett. 20 (2018) 3443-3446. DOI:10.1021/acs.orglett.8b00981 |
| [29] |
Z.M. Tan, X.R. He, K. Xu, et al., ChemSusChem 15 (2022) e202102360. |
| [30] |
P. Peng, Z.H. Zhou, K.F. Hu, et al., Org. Lett. 21 (2019) 6403-6407. DOI:10.1021/acs.orglett.9b02317 |
| [31] |
K. Liu, J.R. Wu, Y.Q. Deng, et al., ChemElectroChem 6 (2019) 4173-4176. DOI:10.1002/celc.201900138 |
| [32] |
K. Liu, S. Tang, T. Wu, et al., Nat. Commun. 10 (2019) 639. DOI:10.1038/s41467-019-08414-8 |
| [33] |
Z.Q. Fan, X.J. Yang, C. Chen, Z.L. Shen, M.C. Li, J. Electrochem. Soc. 164 (2017) G54-G58. DOI:10.1149/2.1561704jes |
| [34] |
C. Chen, P.F. Niu, Z.L. Shen, M.C. Li, J. Electrochem. Soc. 165 (2018) G67-G74. DOI:10.1149/2.0071807jes |
| [35] |
Y.C. Cai, P.F. Niu, Z.L. Shen, M.C. Li, Chem. J. Chin. U. 40 (2019) 2308-2313. DOI:10.7503/cjcu20190330 |
| [36] |
J.B. Henry, R.J. MacDonald, H.S. Gibbad, H. McNab, A.R. Mount, Phys. Chem. Chem. Phys. 13 (2011) 5235-5241. DOI:10.1039/c0cp02645j |
| [37] |
L. Marin, A. Bejan, S. Shova, Dyes Pigment. 175 (2020) 108164. DOI:10.1016/j.dyepig.2019.108164 |
| [38] |
J. Luo, Z. Wan, C. Jia, Chin. Chem. Lett. 27 (2016) 1304-1318. DOI:10.1016/j.cclet.2016.07.002 |
| [39] |
X. Liu, P. Niu, J. Jin, Z.L. Shen, M.C. Li, Electrochim. Acta 331 (2020) 135371. DOI:10.1016/j.electacta.2019.135371 |
| [40] |
J.L. Jin, L.M. Zhao, C. Zhang, et al., J. Electrochem. Soc. 168 (2021) 025504. DOI:10.1149/1945-7111/abe28f |
| [41] |
L.M. Zhao, X. Liu, S.X. Shi, et al., Electrochim. Acta 389 (2021) 138748. DOI:10.1016/j.electacta.2021.138748 |
| [42] |
J. Heinze, A. Rasche, M. Pagels, B. Geschke, J. Phys. Chem. B 111 (2007) 989-997. DOI:10.1021/jp066413p |
| [43] |
M. Goswami, A. Konkel, M. Rahimi, et al., Chem. Eur. J. 24 (2018) 11936-11943. DOI:10.1002/chem.201800730 |
| [44] |
E. Madej, P. Wardman, Radiat. Phys. Chem. 75 (2006) 990-1000. DOI:10.1016/j.radphyschem.2006.01.007 |
| [45] |
Q.R. Huang, Y.C. Li, K.L. Ho, J.L. Kuo, Phys. Chem. Chem. Phys. 20 (2018) 7653-7660. DOI:10.1039/C8CP00533H |
| [46] |
S. Burman, K. Bhattacharya, D. Mukherjee, G. Chandra, BMC Complem. Altern. Altern. M. 18 (2018) 213. DOI:10.1186/s12906-018-2271-0 |
| [47] |
Q. Osmani, H. Hughes, K. Flavin, et al., Anal. Bioanal. Chem. 391 (2008) 1229-1236. DOI:10.1007/s00216-008-1867-5 |
| [48] |
M. Orief, M. Abdel-Rhman, J. Mol. Struct. 1173 (2018) 332-340. DOI:10.1016/j.molstruc.2018.05.107 |
| [49] |
A.M. Erkabaev, T.V. Yaroslavtseva, S.E. Popov, O.V. Bushkova, Vib. Spectrosc. 75 (2014) 19-25. DOI:10.1016/j.vibspec.2014.08.010 |
| [50] |
P.Z. Zhao, K. Wang, Y.Y. Yue, et al., ChemCatChem 12 (2020) 3207-3211. DOI:10.1002/cctc.202000284 |
| [51] |
X. Pan, M. Lamson, J. Yan, K. Matyjaszewski, ACS Macro Lett. 4 (2015) 192-196. DOI:10.1021/mz500834g |
| [52] |
Y. Yu, Z.B. Yue, L.G. Ding, Y. Zhou, H. Cao, ChemistrySelect 4 (2019) 1117-1120. DOI:10.1002/slct.201804056 |
| [53] |
W. Kim, H.Y. Kim, K. Oh, J. Org. Chem. 86 (2021) 15973-15991. DOI:10.1021/acs.joc.1c00873 |