 2023, Vol. 34
2023, Vol. 34 


b Tongliao Center for Disease Control and Prevention, Tongliao 028000, China
Hydrogen peroxide (H2O2) is one of most valuable chemicals and widely used in surgical disinfection, pulp bleaching, water purification, chemical synthesis, etc. [1-5]. The Global Industry Analysts, Inc predicts that the global market consumption of H2O2 will be reach to 6.0 million tons in 2024 [6]. The current industrial method for synthesis H2O2 is the multi-step anthraquinone process, which is an indirect chemical, energy intensive and environmental pollution technique [7]. An alternative and attractive route for H2O2 production is electrochemical oxygen reduction via a two-electron (2e−) pathway, in which oxygen is adsorbed on the electrode surface and converted to H2O2 [8-11]. However, two-electron oxygen reduction reaction (2eORR) is always accompanied by the competition of four-electron (4e−) oxygen reduction reaction (4eORR). In order to trigger two-electron pathway and suppress of subsequent four-electron step for the efficient electrosynthesis of H2O2, the rational design of high active, selective and stable solid catalyst is necessary.
Although noble metals catalysts (NMCs) such as Pd, Au [12-14] and single-atom catalysts (SACs) such as Co-SAC, Mn-SAC [15-18] have been widely used as 2eORR catalysts, the high price of NMCs with scarcity nature, and simultaneously the complicated preparation techniques and poor stability of SACs hider their large-scale applications. Recently, cost-effective metal-free carbon-based materials emerge as alternative candidates for electrochemical synthesis H2O2 from 2eORR [19-26]. With the aim of improving its intrinsic activity, two efficient strategies of surface oxidation [22-24] and heteroatoms (B [27-29], N [30,31], F [32], S [30], etc.) doping are widely explored. These doping atoms extremely change the localized bonding microenvironment, charge state and electronic structures of active site at carbon solids. However, the precise regulation of localized microenvironment around the active site at carbon solid to determine the structure-activity relationship is a still great challenge for metal-free solid site at carbons towards efficient H2O2 electrosynthesis.
Due to the property of metalloid and weak electronegativity, boron (B) site can activate oxygen molecule to trigger the ORR. Moreover, coordination structure of B site can be modulated by C and N localized bonding microenvironment, owing to the fact that B and C can form a homogeneous solid solution with the coexistence of N. In our previous study [33], coordination unsaturated B has been proven to exhibit a better electrochemical nitrogen reduction reaction capability in comparison to the coordination saturated one. And, the different nitrogen activation ability is attributed to the local electron density of B sites. According to the sabatier principle [34-37], an optimum B sites at solid catalyst should bind OOH neither too strongly nor too weakly. Considering the dependency of H2O2 selectivity on the binding energy of reaction intermediate OOH* (ΔGOOH*) reported by previous studies [38,39], we deduce that the fine-tuning of charge state on B site is the key to optimize the ΔGOOH* to thus promote the 2eORR catalytic activity, selectivity and stability duration.
Herein, we focus on the relationship between microenvironment modulation of B site and the electrocatalytic performance of 2eORR. We demonstrate a one-step solid-state reaction between BN and porous C to construct boron-carbon nitrides (BCNs) with abundant chemical bondings of N-B-N, N-B-C and C-B-C. The effects of reaction temperature on the localized bonding microenvironment of B sites and their H2O2 electrosynthesis properties have also been investigated. A selected temperature of 1100 ℃ result in a completely conjunction between BN and C in BCN-1100, and it exhibits an excellent 2eORR performance both in H2O2 selectivity and electrocatalytic stability. The density functional theory (DFT) calculations have also been used to identify the reaction energetics of H2O2 production on different localized structure and charge state of B sites. The BNC2-Gr configuration exhibits a suitable B charge state of 1.89 |e| and optimal OOH* adsorption energy (ΔGOOH*) of 4.24 eV. Therefore, oxygen reduction will undergo two-electron route instead of four-electron one.
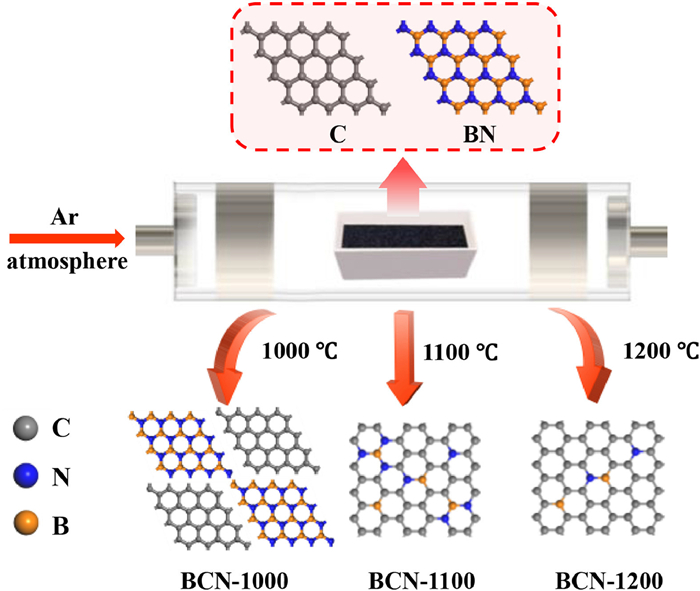
Scheme 1 illustrates vividly the modulation of localized bonding microenvironment on boron (B) site through controlling the temperature of solid-state reaction between the boron nitride (BN) and porous carbon (C). Three temperatures at 1000, 1100 and 1200 ℃ are selected to obtain three boron-carbon nitrides (BCN-1000, BCN-1100 and BCN-1200) with different localized bonding microenvironment at B site. BCN-1000 represents two separate phases of BN and C. BCN-1100 and BCN-1200 indicate the chemical bonding of N-B-C and C-B-C at B site, where the atomic ratio of B and N in BCN-1100 is larger than the one in BCN-1200.

|
Download:
|
| Scheme 1. Schematic illustration for the modulation of localized bonding microenvironment on boron site by carbon and nitrogen at variable temperatures. BCN-1000 representing two phases of BN and C, BCN-1100 and BCN-1200 denoting the B site with chemical bonding of N-B-C and C-B-C. | |
{kind=link}
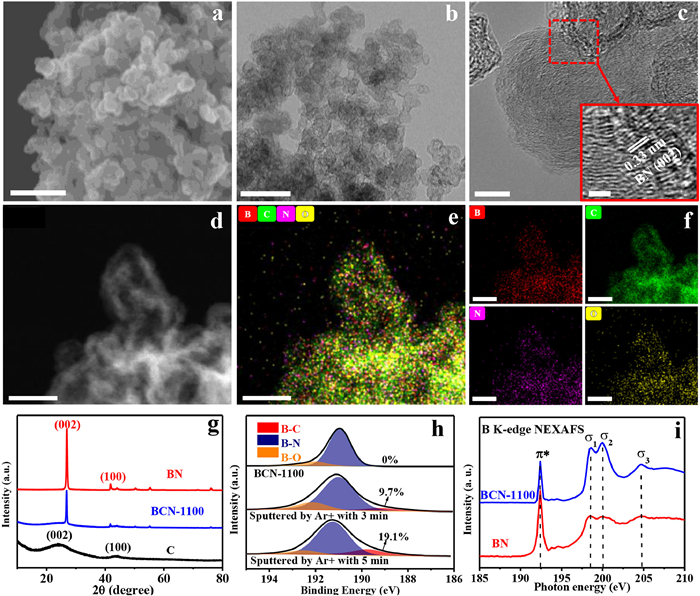
To probe the valid chemical conjunction between BN and C during the solid-state reaction process, we firstly characterized the morphology of BCN-1100 using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Different from the lamellar morphology of BN (Fig. S1a in Supporting information), BCN-1100 and C show similar nanoparticles microstructure (Figs. 1a and b, Fig. S1b in Supporting information). No appearance of lamellar structure in BCN-1100 demonstrates the sufficient solid-state reaction between the crystalline BN lamellar and amorphous C particle. High-resolution transmission electron microscopy (HRTEM) image in Fig. 1c shows the clear lattice diffraction fringes of BCN-1100 in comparison to C-1100 (Fig. S2 in Supporting information), the space of the lattice fringe is 0.33 nm, attributed to the (002) plane of BN where the BN may be covered by the graphite shell. High angle annular dark field (HAADF) and energy dispersive spectroscopy (EDS) mappings (Figs. 1d-f) demonstrate homogeneous elemental distributions of B, C, N and O in BCN-1100.

|
Download:
|
| Fig. 1. Structural characterizations of boron site at BCN-1100. (a) SEM image, scale bar 200 nm. (b) TEM image, scale bar 100 nm. (c) High-resolution TEM image, scale bar 10 nm, inset scale bar 2 nm. (d) HADDF-STEM image, scale bar 20 nm. (e) Elemental mappings for B, C, N and O (merged). (f) Elemental mappings for B, C, N and O (individual). (g) XRD patterns of C, BN and BCN-1100. (h) High resolution B 1s XPS spectra with sputtered ones by argon ion (Ar+) etching at 3 min and 5 min. (i) NEXAFS spectra of B K-edge for BCN-1100 and BN. | |
{kind=link}
Then, we used the X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), near edge X-ray absorption fine structure spectroscopy (NEXAFS) and Raman spectra to explore the localized structure of B site at the BCN-1100. XRD patterns reveal that the hexagonal BN-like lattice for BCN-1100, distinct from the amorphous graphite-like C (Fig. 1g). XPS survey (Fig. S3 and Table S1 in Supporting information) gives the elemental composition in BCN-1100 surface with the element contents of B, C, N and O of 10.78, 78.91, 8.38 and 1.93 at%, respectively. In order to obtain more information on chemical bonds of B site at the BCN-1100, the Ar+ plasma sputtered surface was performed to examine the high-resolution B 1s spectrum. The B 1s spectrum (Fig. 1h and Table S2 in Supporting information) reveal that B-N (191 eV) and B-O (192 eV) existing on the surface. And, the B-C (189.2 eV) emerging in the depth of BCN-1100, with the contents of B-C increasing from 9.7% to 19.1% with the sputtered time from 3 min to 5 min. The same phenomenon can be observed in N 1s spectrum (Fig. S4 and Table S3 in Supporting information), where the peaks located at 398.5 and 399.4 eV are corresponding to N-B and N-C, respectively. The content of N-C increases from 9.2% to 21.4% after Ar+ sputtered 3 min and 24.7% after 5 min. Therefore, the localized bonding microenvironment on B site transforms from pure B-N bonds in BN into B-C and N-C bonds in BCN-1100 with C and N coordination to form variable BN3, BN2C, BNC2 and BC3 sites. NEXAFS analysis of B K-edge shows a sharp B 1s π* resonance at 192.4 eV and three σ* ones at 198.6, 200 and 204.6 eV for BCN-1100, similar to the ones of BN (Fig. 1i). However, compared with BN, BCN-1100 exhibits a significant phenomenon of π* weakening and σ* strengthening, which may be caused by the insertion of B-C into the B-C-N skeleton disordering the symmetry of coordination structure around the B sites. Raman spectra presents the typical defect (D) band and graphite (G) band attributing to the C in BCN-1100, without representative Raman shift for BN. The ID/IG (intensity ratio of D and G) in BCN-1100 is 1.03, which is higher than that of C (0.91), indicating that the N-B-C and C-B-C bonds would increase the defects and disorder structure (Fig. S6 in Supporting information). The Brunauer–Emmett–Teller (BET) analysis determines that the specific surface area of BCN-1100 is 860.8 m2/g, 89.7 times larger than the one of BN (9.6 m2/g) and smaller than that of C (1415.9 m2/g) (Fig. S7 in Supporting information). BCN-1100 shows a mesoporous structure with the pore size distributions between 15 and 25 nm, similar to the one of porous C control (Fig. S8 in Supporting information).
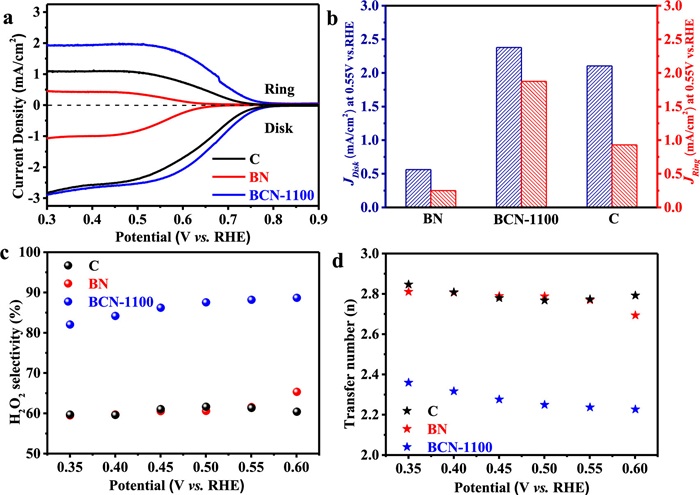
The electrocatalytic two-electron oxygen reduction (2eORR) performances of boron (B) site at BCN-1100 were examined in O2 saturated electrolyte (a solution of 0.1 mol/L KOH) using the rotating ring-disk electrode (RRDE) setup at 1600 rpm. Fig. 2a shows electrochemical polarization results, where the disk electrode (Disk) represents the oxygen reduction current and the ring electrode (Ring) represents the H2O2 oxidation current (collection efficiency is 0.37). The polarization curves show that the BCN-1100 exhibits a higher oxygen reduction current density and an onset potential (defined as the potential achieves disk current density of 0.25 mA/cm2) of 0.74 V vs. reversible hydrogen electrode (RHE), very close to the thermodynamic equilibrium potential for 2eORR (O2 + H2O + 2e− ⇋ HO2− + OH−, 0.75 vs. RHE). The ring current density of BCN-1100 is significantly higher than that of C and BN, indicating that more H2O2 generation could be detected on the disk electrode. To clear depict the effects of B site on the disk currents (JDisk) and ring currents (JRing), we compared the BCN-1100 with two control sample of BN and C in Fig. 2b, where BCN-1100 shows a current density both in JDisk of 2.38 mA/cm2 and JRing of 1.86 mA/cm2, higher than the one of C (2.1 mA/cm2 and 0.92 mA/cm2) and of BN (0.56 mA/cm2 and 0.24 mA/cm2). The B site at BCN-1100 presents a H2O2 selectivity of 89% under potential of 0.55 V vs. RHE (Fig. 2c), one of the highest performance relative to the recently reported non-metal electrocatalysts for 2eORR (Table S4 in Supporting information). And, it also gives an electron transfer number (nRRDE) of 2.2 in Fig. 2d, close to the theoretical ideal 2 electrons required for the reduction from O2 to H2O2. Furthermore, the rotating disk electrode (RDE) voltammograms at different rotation rates were tested to further verify the H2O2 selectivity, an electron transfer number (nRDE) of 2.29 was obtained for BCN-1100, calculated from the Koutechy-Levich equation (K-L equation), very close to nRRDE (Fig. S9 in Supporting information).

|
Download:
|
| Fig. 2. Electrocatalytic two-electron oxygen reduction (2eORR) performances of boron site at BCN-1100. (a) RRDE voltammograms at 1600 rpm in O2-saturated 0.1 mol/L KOH electrolyte. (b) Disk and ring current densities at 0.55 V vs. RHE. (c) H2O2 selectivity. (d) Electron transfer numbers at different potentials. | |
{kind=link}
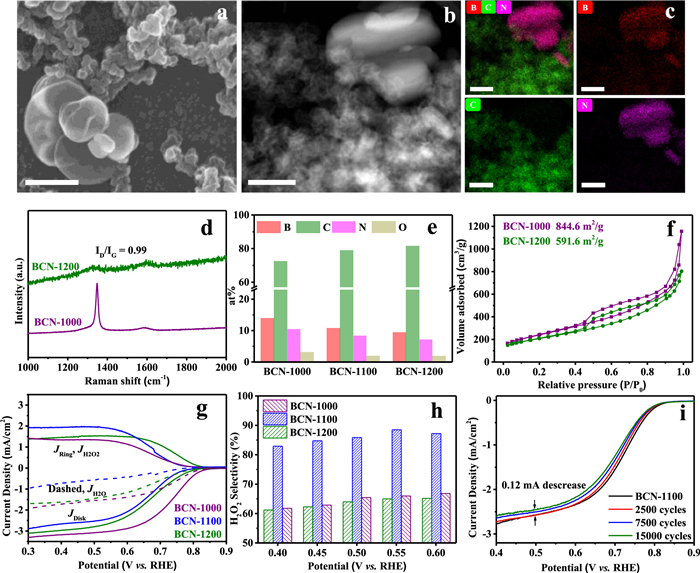
In order to understand the modulation of localized bonding microenvironment at boron site by solid-state reaction temperature, and to clarify the relationships between B site and its electrocatalytic 2eORR performance, we further explored the composition of B sites at BCN-1000 and BCN-1200 and their electrocatalytic properties. No obvious difference in XRD patterns of BCN-1000 and BCN-1200 was observed in comparison to that of BCN-1100 (Fig. S10 in Supporting information). However, the SEM, HADDF and elemental mapping analysis in Figs. 3a-c provide evidences that the BCN-1000 containing BN nanosheets and C nanoparticles, indicating that two phase separation between lamellar BN and particle morphological C remains at 1000 ℃. Raman spectra of BCN-1000 (Fig. 3d) show a sharp BN peak at 1348 cm−1 and two broad C peaks between 1200 cm−1 and 1800 cm−1, which further reveals the mixture of BN and C to constitute the BCN-1000. After the solid-state reaction temperature rising to 1200 ℃, the morphology of BCN-1200 shows no obvious differences in SEM image (Fig. S11 in Supporting information). Raman analysis shows the ID/IG of 0.99 for BCN-1200, slight lower than the one of BCN-1100 indicating that the temperature rising could increase the graphitized carbon in boron-carbon nitride (Fig. 3d). As uptrend of the solid-state reaction temperature, the ratio of C increases 2.61% accompanied by the descending ratios of 1.38% for B and 1.20% for N (no obvious change for O) for BCN-1200 in comparison to that for BCN-1100 (Fig. 3e, Fig. S12 and Table S5 in Supporting information). The specific surface area of 844.6 m2/g for BCN-1000 and of 591.6 m2/g for BCN-1200 are obtained (Fig. 3f). The collapse of pore during high temperature reaction may lead to the decrease of specific surface area of boron-carbon nitride solid, resulting in the decrease of contact area between the catalyst, electrolyte and oxygen molecules.

|
Download:
|
| Fig. 3. Effects of the localized bonding microenvironment at boron site on electrocatalytic 2eORR performances. (a) SEM image of BCN-1000, scale bar 200 nm. (b) HADDF-STEM image of BCN-1000, scale bar 20 nm. (c) Corresponding elemental mappings of B, C and N. (d) Raman spectra. (e) Atomic compositions determined by XPS and (f) N2 adsorption/desorption isotherms. (g) Comparison of RRDE voltammograms at 1600 rpm in O2-saturated 0.1 mol/L KOH electrolyte and (h) H2O2 selectivity at different potentials. (i) Electrochemical stability of BCN-1100. | |
{kind=link}
The 2eORR performances of BCN-1000 and BCN-1200 were investigated in Fig. 3g, where JD is the disk current density, JR (JH2O2) is the ring current density and JH2O is the current density for O2 reduction to H2O: JH2O = JD – JR. BCN-1000 and BCN-1200 exhibit an onset potential of 0.81 V and of 0.78 V (vs. RHE), higher than BCN-1100 demonstrating the competitive four-electron oxygen reduction (4eORR) route occurring in BCN-1000 and BCN-1200 and lower H2O2 selectivity. Moreover, the higher JH2O represents the more four-electron pathway for BCN-1000 and BCN-1200. Then, the ORR currents were normalized by their electrochemical surface area (ECSA), BCN-1100 shows the highest intrinsic activity for electrochemical oxygen reduction (Figs. S13 and S14 in Supporting information). As shown in Fig. 3h, we have plotted the H2O2 selectivity at the potentials between 0.4 V and 0.6 V (vs. RHE) to show the effects of localized bonding microenvironment at boron site. Obviously, BCN-1100 shows a higher H2O2 selectivity than BCN-1000 and BCN-1200 at each potentials, which may due to the higher content of B site with C and N coordination structure in BCN-1100. In addition, BCN-1100 keeps an excellent catalytic stability after a 15000 cycles during CVs test. At the potential of 0.5 V (vs. RHE), the disk electrode only produces a current density drop of 0.12 mA/cm2 (Fig. 3i). The SEM (Fig. S15 in Supporting information) and B 1s XPS spectra (Fig. S16 in Supporting information) of recycled BCN-1100 show no differences with initial fresh BCN-1100. Finally, the H2O2 production and faradaic efficiency (FEH) of BCN-1100 was measured using an H-type cell with chronoamperometry operation and shown in Fig. S18 (Supporting information), where the H2O2 concentration in electrolyte goes up to 10.55 mmol/L and the FEH is maintained up to 80% after 2.5 h. Therefore, the solid-state reaction of BN and C at 1100 ℃ could achieve a balance between high B content and coordinate microstructures at B site in boron-carbon nitride, resulting in an excellent 2eORR activity, selectivity and cycle duration stability.
With the aim of exploring the structure-activity relationship and the 2eORR catalytic process and mechanism at B site, the DFT calculations were performed over several simplified structural models with different B coordination environment based on single-layer carbon structure (Fig. S19 in Supporting information). In addition, the configuration of NC3-Gr (Fig. S20 in Supporting information) was also analyzed for comparison. The BN3-Gr, BN2C-Gr, BNC2-Gr and BC3-Gr structures were used to model the 2eORR reaction pathway:
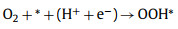
|
(1) |
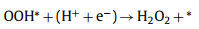
|
(2) |
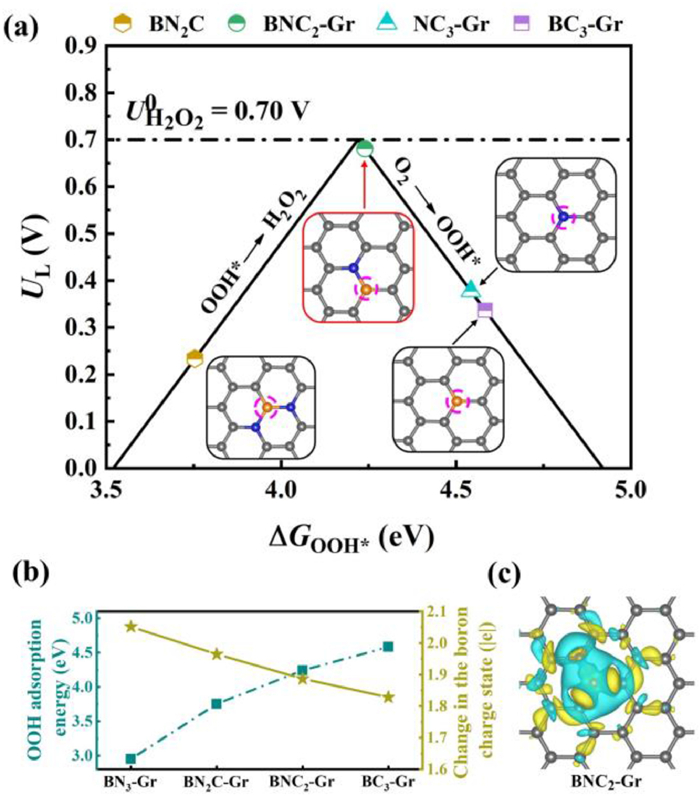
In which, * represents the active sites. The binding energies of the reaction intermediates to the active sites (the B atom in BN3-Gr, BN2C-Gr, BNC2-Gr and BC3-Gr, N atom in NC3-Gr) are calculated and demonstrated in Figs. S19 and S20. Obviously, the overpotential is determined by either the hydrogenation of oxygen or the reduction of OOH* to form H2O2. We then adopted the adsorption energy of OOH* (ΔGOOH*) as a descriptor and plotted the volcano between activity and ΔGOOH* to underline the 2eORR activities of the model structures. The calculated limiting potential (UL), which can be considered as an indicator of activity, is defined as the maximum potential at which the two reaction steps are downhill in free energy. And the theoretical overpotential is defined as the difference between the limiting potential and the equilibrium potential (U = 0.7 V vs. RHE) [23]. In theory, an ideal catalyst should have a ΔGOOH* near 4.22 eV, which provides the highest activity. Fig. 4a shows the calculated dependency relationship of UL and ΔGOOH* for the 2eORR. Among the model B sites, BNC2-Gr has the highest activity for 2eORR with overpotential of 0.02 V and ΔGOOH* of 4.24 eV. While the BN3-Gr is absent in the volcano plot due to the large overpotential (1.27 V) and low ΔGOOH* (2.95 eV). Moreover, the catalytic activity of N site in NC3-Gr is not ideal, yielding an overpotential of 0.32 eV.

|
Download:
|
| Fig. 4. Theoretical analysis of the structure and charge state for the localized bonding microenvironment at boron site. (a) Calculated relationship between structure and catalytic activity for the production to H2O2 via 2eORR. The limiting potential is plotted as a function of ΔGOOH*, and the equilibrium potential is shown as the dashed black line. (b) Influences of localized bonding microenvironment on the OOH* adsorption energies and relevant charge states at the boron site. (c) Differential charge densities of the BNC2-Gr model with highest 2eORR activity. Cyan and yellow isosurfaces (±0.002 Bohr−3) donating electron losing and gaining, respectively. | |
{kind=link}
Interestingly, Figs. 4a and b show that ΔGOOH* can be adjusted by regulating the coordination microenvironment of B site, and the 2eORR catalytic performance will be subsequently obtained. With the increase of coordinated N atoms and the decline of coordinated C atoms at B site, ΔGOOH* shows a rising tendency and gets very close to the optimal value of H2O2 production in the microenvironment of BNC2. Further microenvironment modulation to BC3 will result in the increasing of ΔGOOH* to 4.54 eV, and yielding the reduced 2eORR performance with an overpotential of 0.32 V. These effects of coordination bonding microenvironment on the ΔGOOH* can be demonstrated by the charge state of B site. Owing to the lower electronegativity of B (2.55), the charge state becomes positive when it is coordinated to the N and C atoms (Figs. 4b and c). Notably, the charge state of B decreases almost linearly when the coordination microenvironment is regulated by increasing the coordinated N atom and declining the coordinated C atom, owing to the higher electronegativity of N (3.04) with respect to C (2.55). With the charge state at B site declines, the ΔGOOH* shows an increasing tendency. Only if the adsorption energy of OOH* is suitable (i.e., neither too high nor too low), the excellent 2eORR performance can be achieved. This can also be demonstrated by the crystal orbital Hamilton population (COHP) analyses. The theoretical value of ICOHP can express the bonding strength based on counting the energy-weighted population of wave functions between two atomic orbitals [40]. Values of ICOHP corresponding to BN3-Gr, BN2C-Gr, BNC2-Gr and BC3-Gr configurations are listed in the order of −8.29, −7.62, −7.35 and −6.56. The larger absolute value of ICOHP means a stronger binding. It is shown that the B site in BN3-Gr present strong binding toward OOH*, and Eq. 2 is the rate-determining step. As for the BC3-Gr configuration with weak bonding strength, it locates at the right side of the volcano plot, hence, Eq. 1 is the rate-determining one [41]. The BNC2-Gr configuration with optimal binding strength appears closed to the top of the volcano and thus exhibiting the outstanding 2eORR performance. In these models we investigated here, the BNC2-Gr model with the highest activity exhibits the B charge state of 1.89 |e|, which could be considered as the optimum B charge state for 2eORR. Therefore, the modulated charge state of B site through the coordination microenvironment regulating is the underlying origin of the varied 2eORR performance here, and it would be a rational strategy for obtaining metal-free electrocatalysts with B site towards high 2eORR activity.
In summary, we provide a metal-free solid boron site to realize an efficient selective electrocatalytic H2O2 production. The C and N localized bonding microenvironment at boron-carbon nitride solid can be modulated through solid-state reaction at different temperatures. The chemical bonding of N-B-C and C-B-C in configurations of BN3, BN2, BNC and BC coordination bonding microenvironment affects the charge state at B site and corresponding adsorption energy of OOH* (ΔGOOH*) in rate-determining step and its electrocatalytic 2eORR activity for H2O2 production. This work paves the pathway for rational design of metal-free solid site by charge state modulation in boosting the green electrochemical synthesis of hydrogen peroxide (H2O2) via two-electron oxygen reduction.
Declaration of competing interestWe declared no conflict of interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 22161036, 11904187, 21961024 and 21961025), Natural Science Foundation of Inner Mongolia (Nos. 2018JQ05 and 2019BS02007), Incentive Funding from Nano Innovation Institute (NII) of Inner Mongolia Minzu University, the Inner Mongolia Autonomous Region Funding Project for Science & Technology Achievement Transformation (Nos. CGZH2018156 and 2019GG261) and Doctoral Scientific Research Foundation of Inner Mongolia Minzu University (Nos. BS437 and BS480).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.06.019.
| [1] |
C. Xia, Y. Xia, P. Zhu, L. Fan, H. Wang, Science 366 (2019) 226-231. DOI:10.1126/science.aay1844 |
| [2] |
C. Xia, J.Y. Kim, H. Wang, Nat. Catal. 3 (2020) 605-607. DOI:10.1038/s41929-020-0486-1 |
| [3] |
X. Shi, S. Back, T.M. Gill, S. Siahrostami, X. Zheng, Chem 7 (2020) 38-63. |
| [4] |
S.C. Perry, D. Pangotra, L. Vieira, et al., Nat. Rev. Chem. 3 (2019) 442-458. DOI:10.1038/s41570-019-0110-6 |
| [5] |
X. He, H. Shang, C. Wang, et al., Chin. Chem. Lett. 32 (2021) 3377-3381. DOI:10.1016/j.cclet.2021.04.028 |
| [6] |
X. Zhang, Y. Xia, C. Xia, H. Wang, Trends Chem. 2 (2020) 942-953. DOI:10.1016/j.trechm.2020.07.007 |
| [7] |
J.M. Campos Martin, G. Blanco Brieva, J.L. Fierro, Angew. Chem. Int. Ed. 45 (2006) 6962-6984. DOI:10.1002/anie.200503779 |
| [8] |
S. Yang, A. Verdaguer-Casadevall, L. Arnarson, et al., ACS Catal. 8 (2018) 4064-4081. DOI:10.1021/acscatal.8b00217 |
| [9] |
Y. Jiang, P. Ni, C. Chen, et al., Adv. Energy Mater. 8 (2018) 1801909. DOI:10.1002/aenm.201801909 |
| [10] |
Y. Pang, H. Xie, Y. Sun, M.M. Titirici, G.L. Chai, J. Mater. Chem. A 8 (2020) 24996-25016. DOI:10.1039/D0TA09122G |
| [11] |
Y. Sun, L. Han, P. Strasser, Chem. Soc. Rev. 49 (2020) 6605-6631. DOI:10.1039/D0CS00458H |
| [12] |
M. Traube, Ber. Kgl. Akad. Wiss. Berlin 1041 (1887) 185. |
| [13] |
S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, et al., Nat. Mater. 12 (2013) 1137-1143. DOI:10.1038/nmat3795 |
| [14] |
J.S. Jirkovsky, I. Panas, E. Ahlberg, et al., J. Am. Chem. Soc. 133 (2011) 19432-19441. DOI:10.1021/ja206477z |
| [15] |
E. Jung, H. Shin, B.H. Lee, et al., Nat. Mater. 19 (2020) 436-442. DOI:10.1038/s41563-019-0571-5 |
| [16] |
R. Shen, W. Chen, Q. Peng, et al., Chem 5 (2019) 2099-2110. DOI:10.1016/j.chempr.2019.04.024 |
| [17] |
C. Tang, Y. Jiao, B. Shi, et al., Angew. Chem. Int. Ed. 132 (2020) 9256-9261. DOI:10.1002/ange.202003842 |
| [18] |
K. Jiang, S. Back, A.J. Akey, et al., Nat. Commun. 10 (2019) 3997. DOI:10.1038/s41467-019-11992-2 |
| [19] |
D. San Roman, D. Krishnamurthy, R. Garg, et al., ACS Catal. 10 (2020) 1993-2008. DOI:10.1021/acscatal.9b03919 |
| [20] |
L. Li, C. Tang, Y. Zheng, et al., Adv. Energy Mater. 10 (2020) 2000789. DOI:10.1002/aenm.202000789 |
| [21] |
Y.J. Sa, J.H. Kim, S.H. Joo, Angew. Chem. Int. Ed. 58 (2019) 1100-1105. DOI:10.1002/anie.201812435 |
| [22] |
H.W. Kim, M.B. Ross, N. Kornienko, et al., Nat. Catal. 1 (2018) 282-290. DOI:10.1038/s41929-018-0044-2 |
| [23] |
Z. Lu, G. Chen, S. Siahrostami, Z. Chen, et al., Nat. Catal. 1 (2018) 156-162. DOI:10.1038/s41929-017-0017-x |
| [24] |
G.F. Han, F. Li, W. Zou, et al., Nat. Commun. 11 (2020) 2209. DOI:10.1038/s41467-020-15782-z |
| [25] |
B. Wang, X. Cui, J. Huang, R. Cao, Q. Zhang, Chin. Chem. Lett. 29 (2018) 1757-1767. DOI:10.1016/j.cclet.2018.11.021 |
| [26] |
H.J. Zhang, J. Geng, C. Cai, et al., Chin. Chem. Lett. 32 (2021) 745-749. DOI:10.1016/j.cclet.2020.05.002 |
| [27] |
S. Chen, Z. Chen, S. Siahrostami, et al., J. Am. Chem. Soc. 140 (2018) 7851-7859. DOI:10.1021/jacs.8b02798 |
| [28] |
G. Coria, T. Pérez, I. Sirés, J.L. Nava, J. Electroanal. Chem. 757 (2015) 225-229. DOI:10.1016/j.jelechem.2015.09.031 |
| [29] |
Y. Xia, X. Zhao, C. Xia, et al., Nat. Commun. 12 (2021) 4225. DOI:10.1038/s41467-021-24329-9 |
| [30] |
V. Perazzolo, C. Durante, R. Pilot, et al., Carbon 95 (2015) 949-963. DOI:10.1016/j.carbon.2015.09.002 |
| [31] |
D. Iglesias, A. Giuliani, M. Melchionna, et al., Chem 4 (2018) 106-123. DOI:10.1016/j.chempr.2017.10.013 |
| [32] |
K. Zhao, Y. Su, X. Quan, et al., J. Catal. 357 (2018) 118-126. DOI:10.1016/j.jcat.2017.11.008 |
| [33] |
Y. Yang, Y. Wang, X. Wang, et al., Adv. Mater. Interfaces (2022) 2101842. |
| [34] |
F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, et al., Phys. Rev. Lett. 99 (2007) 016105. DOI:10.1103/PhysRevLett.99.016105 |
| [35] |
Y. Wang, H. Su, Y. He, et al., Chem. Rev. 120 (2020) 12217-12314. DOI:10.1021/acs.chemrev.0c00594 |
| [36] |
D. He, L. Cao, L. Feng, et al., Chin. Chem. Lett. 33 (2022) 4781-4785. DOI:10.1016/j.cclet.2022.01.006 |
| [37] |
T. Tang, Z. Wang, J. Guan, Adv. Funct. Mater. (2022) 2111504. |
| [38] |
S. Siahrostami, G.L. Li, V. Viswanathan, J.K. Norskov, J. Phys. Chem. Lett. 8 (2017) 1157-1160. DOI:10.1021/acs.jpclett.6b02924 |
| [39] |
V. Viswanathan, H.A. Hansen, J.K. Norskov, J. Phys. Chem. Lett. 6 (2015) 4224-4228. DOI:10.1021/acs.jpclett.5b02178 |
| [40] |
Z. Liu, D. Li, S. Wei, et al., Inorg. Chem. 56 (2017) 7494-7500. DOI:10.1021/acs.inorgchem.7b00980 |
| [41] |
H. Shao, Q. Zhuang, H. Gao, et al., Inorg. Chem. Front. 8 (2021) 173-181. DOI:10.1039/D0QI01089H |