 2023, Vol. 34
2023, Vol. 34 


b College of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou 350122, China;
c School of Pharmacy, Tongji-Rongcheng Center for Biomedicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
Targeting immune checkpoint pathway is a promising new anticancer therapy. Immunotherapy can stimulate or mobilize the human immune system to kill tumor cells rather than directly kill tumor cells [1-3]. Encouraged by the approval of ipilimumab, a growing number of immune checkpoint inhibitors are under intense development. Among them, the development of PD-1/PD-L1 immune checkpoint inhibitors is the most popular [4-7]. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1/PD-L1 monoclonal antibody has shown great anti-tumor efficacy in various kinds of solid tumors [8]. However, a number of immune-related adverse reactions with fatal consequences of monoclonal antibodies have been reported in these years. Although PD-1/PD-L1 is the most popular small molecule immunotherapy pathway, no small molecule inhibitors have been approved by the FDA. Compared with monoclonal antibodies, small molecule drugs show great advantages, such as oral administration possibility, stability, non-immunogenicity and membrane permeability [9]. Bristol-Myers Squibb disclosed the first patent of biphenyl immunomodulators in 2015 [10]. Since then, many scientific research institutions have modified these structures of diphenyl immunomodulators in order to obtain PD-1/PD-L1 inhibitors with better activity, such as BMS-37 [11], NP19 [12], NB [13] and I-2-A (Fig. S1 in Supporting information) [14].
The idea of proteolysis-target chimera (PROTAC) has gradually entered the field of drug development, which uses the mechanism of ubiquitination to degrade the target protein to achieve the inhibition of the target protein. Traditional small molecules and antibodies are driven by occupation, and this mode of action requires a high concentration of inhibitors or monoclonal antibodies to occupy the active site of the target and block the transduction of downstream signal pathways. PROTAC is event-driven and mediates the degradation of target proteins rather than affecting the function of proteins. At the same time, the target protein can be labeled by ubiquitin, which in theory can be used over and over again, so the catalytic dose can come into play [15]. On the whole, PROTAC is a novel strategy for removing unwanted proteins through the ubiquitin-proteasome system [16].
Recent studies have shown that PD-L1 protein is subject to ubiquitin-mediated proteasome degradation [17,18]. Therefore, it seems feasible to design a novel PD-L1 small molecule degradant based on PROTAC technology. Some studies have pointed out that the two benzene ring structures of biphenyl compounds are located in the deep hydrophobic channel-like pocket of PD-L1 homodimer (Fig. 1A) [19]. Therefore, in this article, we selected the four biphenyl compounds described in Fig. S1 as PD-L1 ligands, and designed PROTAC molecules with different chain lengths or E3 (CRBN, VHL, cIAP, MDM2) ligands [20], in order to improve the degradation activity of PD-L1 degrading agent and provide a new immunotherapy strategy for cancer.

|
Download:
|
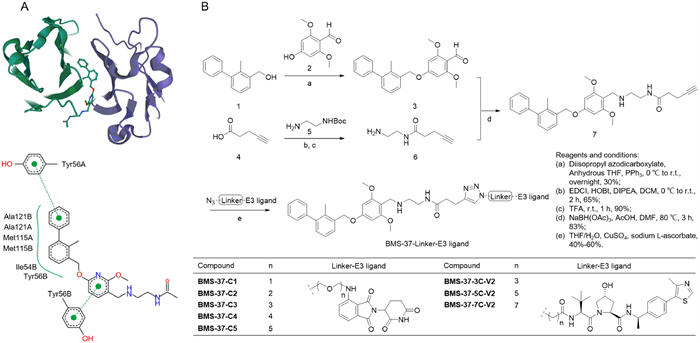
| Fig. 1. The design and synthesis of title compounds. (A) PD-L1 inhibitor and interaction with PD-L1 dimer. (B) The synthesis of BMS-37-linker-E3 ligand. | |
{kind=link}
Firstly, in order to determine the reasonable linker derivatization sites, PoseView (https://proteins.plus/) was used to analyze the interaction between biphenyl compound and PD-L1 homodimer (Fig. 1A). The diphenyl moiety is located inside the binding pocket and forms a π-π stacking with Tyr56C, in addition, it also forms extensive hydrophobic interaction with Met115C, Met115D, Ala121C, Ala121D, Ile54D and Tyr56D. The pyridine ring at the outlet of the pocket is formed a π-π stacking with Tyr56D. The carbonyl group of terminal acetamide forms a hydrogen bond with Lys124C. Based on the above conclusions, terminal acetamide was selected as the connecting site and Linkers were connected through amide condensation reaction (PDB ID: 5J89).
We next synthesized 21 PD-L1 PROTACs based on different PD-L1 ligands (Table S1 in Supporting information). Eight PROTACs with BMS-37 as PD-L1 ligand were synthesized according to Fig. 1B. Raw material 1 was etherified with 4‑hydroxy-2,6-dimethoxybenzaldehyde to obtain intermediate 3. Then NaBH(OAc)3-mediated reductive amination reaction converted intermediate 3 into intermediate 7. Finally, N3-linker-E3 ligand compounds with different lengths were connected with 7 by click reaction, respectively. Schemes S1-S3 (Supporting information) showed the synthesis of the remaining compounds.
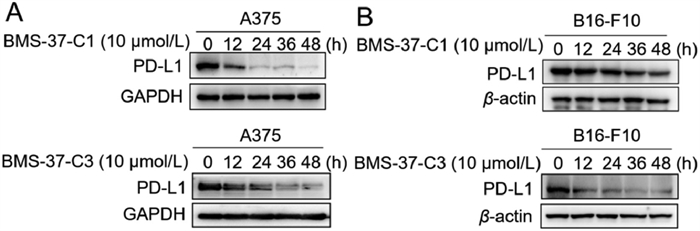
Western blotting (WB) experiments were then used to assay the degradation efficiency of the synthesized compounds in B16-F10 and A375 cell lines. The experimental results demonstrated that BMS-37-C3 and BMS-37-C1 exerted the most efficient impact on the degradation of PD-L1 protein in the A375 cell line. Both compounds started to affect the expression of PD-L1 protein at 0.3 µmol/L. BMS-37-C2 was also effective in the degradation of PD-L1, but its degradation effect was weaker than that of BMS-37-C3. BMS-37-C4 and BMS-37-C5 did not affect the level of PD-L1 protein in the A375 cell line (Fig. S2A in Supporting information).
Furthermore, BMS-37-C3 and BMS-37-C1 inhibited PD-L1 more effectively than other drugs in the B16-F10 cell line (Figs. S2B and S3 in Supporting information). No significant degradation of PD-L1 was observed in the combination of non-BMS series compounds and different E3 ligands (Figs. S5-S7 in Supporting information). So BMS-37-C3 and BMS-37-C1 were chosen in the subsequent experiments. To further demonstrate the degradation kinetics of synthesized PROTACs, time-dependent experiments were carried out. From the results, BMS-37-C3 showed the best degradation ability at 48 h in both cell lines (Fig. 2 and Fig. S4 in Supporting information). Moreover, the growth inhibition effect of the above compounds was evaluated (Table S2 and Fig. S8 in Supporting information).

|
Download:
|
| Fig. 2. BMS-37-C3 and BMS-37-C1 degraded PD-L1 in a time-dependent manner in both A375 cells (A) and B16-F10 cells (B). | |
{kind=link}
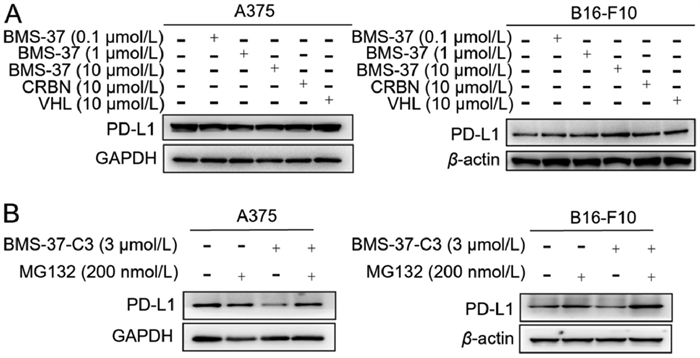
The impact of BMS-37, CRBN, and VHL ligand on PD-L1 expression was further investigated to confirm if the reduction in PD-L1 protein produced by BMS-37-C3 is a PROTAC-mediated degradation. The results demonstrated that the administration of the same dose concentrations revealed that BMS-37, CRBN, and VHL ligand did not affect PD-L1 expression, suggesting that BMS-37-C3 achieved inhibition of PD-L1 expression through a degradation pathway (Fig. 3A). To further verify that the degradation of proteins in cells by BMS-37-C3 functions via the ubiquitination pathway, the effect of BMS-37-C3 was assayed by introducing the proteasome inhibitor MG132. The inhibitory effect of BMS-37-C3 on PD-L1 disappeared after the intervention by giving MG132 (Fig. 3B). This experiment demonstrated that this compound could degrade PD-L1 through the ubiquitination pathway.

|
Download:
|
| Fig. 3. Exploration of specific degradation pathways. (A) Effect of BMS-37, CRBN and VHL on PD-L1 expression in A375 and B16-F10 cells. (B) MG132 reverses the inhibitory effect of BMS-37-C3 on PD-L1 in A375 and B16-F10 cells. | |
{kind=link}
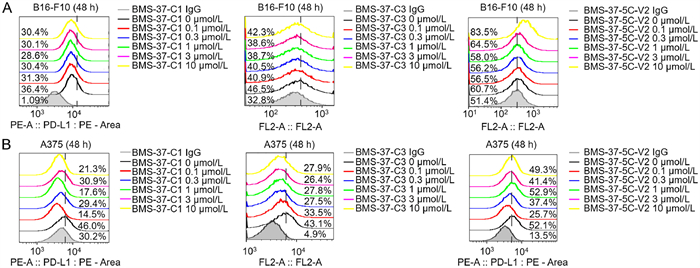
To determine whether the compounds could degrade PD-L1 on the cell surface, flow cytometry was used to detect PD-L1 protein levels on the cell membrane. A375 and B16-F10 cells were chosen for screening the degradation efficiency of the synthesized PROTAC molecules. Flow cytometry assay revealed that BMS-37-C1 at 1 µmol/L reduced cell surface PD-L1 expression to 28.6% when compared with 36.4% of the DMSO-treatment group (Fig. 4). BMS-37-C3 at 1 µmol/L reduced cell surface PD-L1 expression to 38.7% when compared with 46.5% of the DMSO-treatment group (Fig. 4). Other compounds, such as BMS-37–5C-V2, however, did not show statistically significant effects on the degradation of PD-L1 protein. In all, in both B16-F10 and A375 cells, compounds decreased the expression of PD-L1 on cell surface between 0.1 µmol/L and 1 µmol/L, but HOOK effect occurred at 3 µmol/L to 10 µmol/L.

|
Download:
|
| Fig. 4. Cell-surface expression of PD-L1 was examined by flow cytometry. (A) The expression levels of PD-L1 protein were determined in B16-F10 by flow cytometry. (B) The expression levels of PD-L1 protein were determined in A375 by flow cytometry. | |
{kind=link}
The above experiments demonstrated that compound BMS-37-C3 could reduce the expression level of PD-L1 through the ubiquitination pathway, indicating that BMS-37 can successfully degrade PD-L1 after modification by PROTAC strategy. However, the specific ability of BMS-37-C1 and BMS-37-C3 still needs to be reflected in the ability to increase T cell efficiency. T cell co-culture with tumor cells is a model that can respond to the killing effect of T cells on tumor cells [21]. PD-L1 can bind to PD-1 on T cells and conduct immunosuppressive signals, thus inhibiting the activity of effector T cells from hindering their killing of tumor cells. Therefore, BMS-37-C1 and BMS-37-C3 can be introduced to test whether they can affect T-cell killing.
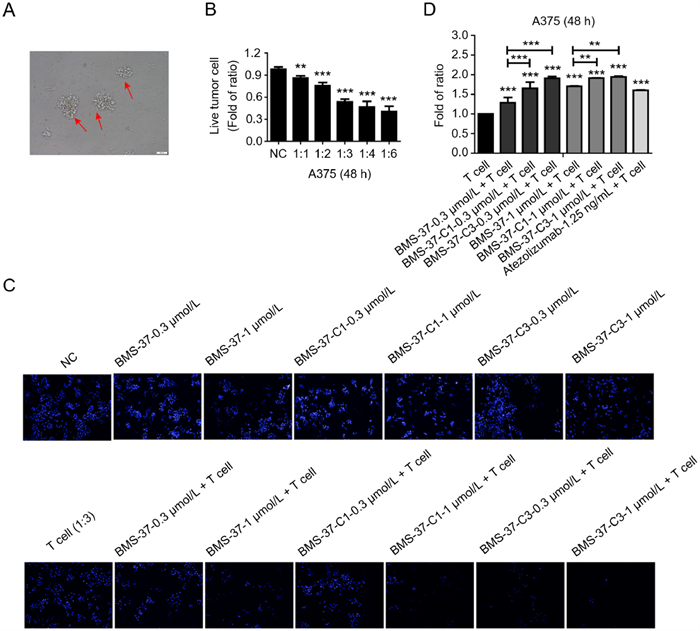
The morphology of activated T cells after stimulation was shown in Fig. 5A. T cells could kill about 50% of tumor cells when the ratio of tumor cells and T cells was 1:3 in co-culture. When the corresponding compounds were added and co-cultured with T cells, BMS-37-C1 and BMS-37-C3 enhanced T cell killing ability to A375 between 0.3 and 1.0 µmol/L in a concentration-dependent manner (Figs. 5B and C). As can be seen from Fig. 5D, the antitumor immune effect of synthetic PROTACs was stronger than the BMS-37 and Atezolizumab.

|
Download:
|
| Fig. 5. The activity of synthesized PD-L1 PROTACs was evaluated by T cell co-culture killing assay. (A) Microscopic view of stimulated T cells after primary culture. (B) The exploration of co-culture ratio of T cells and tumor cells. (C) Cytotoxic effect of BMS-37, BMS-37-C1, BMS-37-C3 on A375 cells after co-culture with T cells. (D) Statistical graph of T cell killing activity of BMS-37, BMS-37-C1, BMS-37-C3 and Atezolizumab. | |
{kind=link}
In order to further confirm the target engagement of these synthesized PD-L1 PROTACs, microscale thermophoresis (MST) was employed to assay the binding affinity between PD-L1 and three representative PROTACs, BMS-37-C1 (CRBN based), BMS-37-C3 (CRBN based), and BMS-37–5C-V2 (VHL based). As shown in Figs. S9 and S10 (Supporting information), the equilibrium dissociation constants Kd of BMS-37, BMS-37-C1, BMS-37-C3, and BMS-37–5C-V2 were 0.12 ± 0.02 nmol/L, 28.70 ± 4.94 µmol/L, 1.61 ± 0.37 µmol/L and 9.10 ± 2.11 µmol/L, respectively. It was further demonstrated that synthetic PROTACs can bind to PD-L1 protein, recruit E3 ligase, and degrade protein through the proteasome pathway.
In this study, a series of PD-L1 PROTAC degraders were synthesized based on four small molecule inhibitors of PD-L1. Two previous studies have reported the synthesis of PROTACs by using BMS-1198 and BMS-37 as PD-L1 inhibitors. Only CRBN was used as ligase system [21,22], while in this paper, a variety of inhibitors of PD-L1 were selected to synthesize PROTACs, such as BMS-37 [11], NP19 [12], NB [13] and I-2-A [14]. Different ligase systems, such as VHL, CRBN, MDM2 and cIAP E3 ligase system were selected. We connected target protein ligand and linker-E3 ligands by click reaction, and applied PEG-linker with different length. The protein degradation level of PD-L1 was detected in different cancer cell lines by intracellular western blot, and the killing ability of T cells on tumor cells was examined in a co-culture model of A375 and T cells. Compared with the previous reported two PD-L1 PROTACs, we systematically studied the effect of BMS-37, BMS-37-C1 and BMS-37-C3 on the killing ability of T cells, and Atezolizumab, a marketed monoclonal antibody (mAb) was used as a positive control. Cell-surface expression of PD-L1 was examined by flow cytometry to evaluate the degradation effect of BMS-37-C1, BMS-37-C3 and BMS-37–5C-V2. In addition, this study showed that the PROTACs of BMS-37 combined with VHL ligands had a certain degradation effect on PD-L1 in dose and time dependent manner. PROTACs of BMS-37 combined with VHL ligands were first reported here with potent degradation effects of PD-L1. In general, this study found a series of strong and effective PD-L1 PROTACs, which had effects in immunotherapy and can enhance the killing ability of T cells.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (NSFC, Nos. 82141216, 81773637 and 81903863), the National Mega-project for Innovative Drugs (No. 2019ZX09721001-004-007, China), the Chunhui Program-Cooperative Research Project of the Ministry of Education, the Natural Science Foundation of Hubei Province (No. 2020CFB642), the Liaoning Province Natural Science Foundation (No. 2020-MZLH-31), and Shenyang Young and Middle-aged Innovative Talents Support Program (No. RC210446). The Center of Analysis and Testing of Huazhong University of Science and Technology is gratefully acknowledged for the characterization of the new compounds.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107762.
| [1] |
O.J. Finn, Ann. Oncol. 23 (2012) ⅷ6-ⅷ9. DOI:10.1093/annonc/mds256 |
| [2] |
D.J. Herzyk, H.G. Haggerty, AAPS J. 20 (2018) 28-39. DOI:10.1208/s12248-017-0184-3 |
| [3] |
Y.Y. Hu, L. Lin, Z.P. Guo, et al., Chin. Chem. Lett. 32 (2021) 1700-1774. |
| [4] |
V.K. Sondak, K.S. Smalley, R. Kudchadkar, et al., Nat. Rev. Drug Discov. 10 (2011) 411-412. DOI:10.1038/nrd3463 |
| [5] |
O. Hamid, C. Robert, A. Daud, et al., N. Engl. J. Med. 369 (2013) 134-144. DOI:10.1056/NEJMoa1305133 |
| [6] |
E.B. Garon, N.A. Rizvi, R. Hui, et al., N. Engl. J. Med. 372 (2015) 2018-2028. DOI:10.1056/NEJMoa1501824 |
| [7] |
B.C. Pestalozzi, D. Zahrieh, E. Mallon, et al., J. Clin. Oncol. 26 (2008) 3006-3014. DOI:10.1200/JCO.2007.14.9336 |
| [8] |
Y.M. Weng, M. Peng, M.X. Hu, et al., OncoTargets Ther. 11 (2018) 7529-7542. DOI:10.2147/ott.s167865 |
| [9] |
J.Y. Zhang, Y.Y. Yan, J.J. Li, et al., Front. Pharmacol. 11 (2020) 722-736. DOI:10.3389/fphar.2020.00722 |
| [10] |
L.S. Chupak, X. Zheng, Patent, WO2015034820, 2015.
|
| [11] |
K. Guzik, K.M. Zak, P. Grudnik, et al., J. Med. Chem. 60 (2017) 5857-5867. DOI:10.1021/acs.jmedchem.7b00293 |
| [12] |
B. Cheng, Y. Ren, X. Niu, et al., J. Med. Chem. 63 (2020) 8338-8358. DOI:10.1021/acs.jmedchem.0c00574 |
| [13] |
J. Li, L. Wu, W. Yao, Patent, WO2017087777, 2017.
|
| [14] |
Z. Shen, X. Sun, Y. Yuan, Patent, CN112724148, 2021.
|
| [15] |
I. Hiroyuki, J. Liu, W.Y. Wei, et al., Acta Materia Medica 1 (2022) 24-41. |
| [16] |
J. Salami, C.M. Crews, Science 355 (2017) 1163-1167. DOI:10.1126/science.aam7340 |
| [17] |
R.P. Wurz, V.J. Cee, et al., J. Med. Chem. 62 (2019) 445-447. DOI:10.1021/acs.jmedchem.8b01945 |
| [18] |
Y. Li, J. Yang, A. Aguilar, et al., J. Med. Chem. 62 (2019) 448-466. DOI:10.1021/acs.jmedchem.8b00909 |
| [19] |
K.M. Zak, P. Grudnik, K. Guzik, et al., Oncotarget 7 (2016) 30323-30335. DOI:10.18632/oncotarget.8730 |
| [20] |
S.L. Paiva, C.M. Crews, Curr. Opin. Chem. Biol. 50 (2019) 111-119. DOI:10.1016/j.cbpa.2019.02.022 |
| [21] |
B. Cheng, Y. Ren, H. Cao, et al., Eur. J. Med. Chem. 199 (2020) 112377. DOI:10.1016/j.ejmech.2020.112377 |
| [22] |
Y. Wang, Y. Zhou, S. Cao, et al., Bioorg. Chem. 111 (2021) 10. DOI:10.1117/12.2577847 |