 2023, Vol. 34
2023, Vol. 34 


Cisplatin [1], carboplatin [2] and loplatin [3] belong to platinum agents, among which cisplatin is the representative of the first generation of platinum agents, while carboplatin and loplatin are the representatives of the second and the third generation of platinum agents, respectively. Although all three agents are broad-spectrum antitumor drugs, cisplatin has the broadest antitumor spectrum and has been widely used for the treatment of a variety of malignant tumors, including head-neck squamous cell carcinoma [4], ovarian cancer [5], non-small cell lung cancer (NSCLC) [6], breast cancer [7], etc. The meta-analyses suggested that cisplatin had 11% higher survival advantage over carboplatin in the treatment of solid tumors like lung cancer [6]. Accordingly, cisplatin has been recommended to clinical uses more often. However, cisplatin chemoresistance often occurs in the clinical treatment [8]. Cisplatin resistance may derive from the acquired resistance by pre-target resistance [9,10], on-target resistance [11], post-target resistance [12] and off-target resistance [13]. Therefore, how to overcome the chemoresistance remains a scientific issue to solve.
The evidence has shown that the chemoresistance of cisplatin is closely related to biological factors in the body such as glutathione (GSH), methionine, metallothionein [14]. Among all these factors, glutathione is a crucial cause for the loss of efficacy in the treatment of tumors. GSH is a low molecular weight peptide that is abundant in cancer cells. As a free radical scavenger, it plays a vital role in maintaining the balance of cell oxidation [15,16]. In the body, GSH often undergoes various reactions in cells through the actions of γ-glutamylcysteine synthetase and glutathione synthetase [17]. Studies have found that GSH is involved in the pre-target resistance of cisplatin. GSH content inside the drug-resistant cancer cells can be 13–50 fold higher compared with that in drug sensitive cells. It is due to the fact that, in the presence of high concentrations of GSH and GSH transferase in cancer cells, Cl− ion in cisplatin can be easily occupied by the sulfydryl (-SH) in GSH. Accordingly, cisplatin would lose the active site of the drug, leading to the inactivation of cisplatin [18].
To solve the issue of cisplatin inactivation, scientists have tried many methods, such as developing prodrugs [19], changing the way of drug delivery [20], and constructing nanocarriers [21]. Prodrug represents a compound that is inactive or less active in vitro by modifying the chemical structure of a drug, but transforms and releases the active drug to exert its efficacy in vivo. For example, platinum(Ⅳ) complexes are often used as prodrugs of cisplatin or its derivatives. Other methods such as changing the route of administration can increase the local delivery and retention of the drug. In particular, nanoparticle drug delivery system exhibits a broad development prospect. Nanoparticles can circumvent the disadvantages of free platinum drug, and ideal nanocarriers can effectively encapsulate the drug and prevent the interaction of platinum from degradation in delivery process [22,23]. However, regular platinum nanoparticle may still face the glutathione depletion in the cancer cells due to the existence of rich glutathione concentration in cancer cells.
Polymer nanoparticles refer to nanoparticles formed by polymer compounds. Commonly used polymer nanoparticle materials consist of synthetic polymers, and natural polymer materials such as polyaspartic acid (Pasp), hyaluronic acid (HA). Pasp has a side chain shuttle group, and hence has unique characteristics of biodegradability, dispersing function and chelating function [24,25]. HA is an immunoneutral linear polysaccharide ubiquitously in the human body. It consists of alternating units of a repeating disaccharide, β-1,4-d-glucuronic acid-β-1,3-N-acetyl-d-glucosamine [26,27]. Through various chemical reactions, Pasp and HA can be modified to alter the hydrophobicity and bioactivity properties, and accordingly, they have been widely used in various polymer block products [28]. Therefore, their versatility of polymer synthesis and processing holds great promise for applications in tissue engineering and drug delivery [29,30].
Here, we develop a novel kind of disulfide switchable nanoparticles complexing cisplatin (switch NPs-cisplatin). In this nano-delivery system, HA (10 kDa), Pasp and disulfide bond compound (cystamine) were used as the basic materials to synthesize a disulfide-rich polymer (HA-ss-Pasp). Cisplatin was covalently bonded to HA-ss-Pasp polymer to form switch NPs-cisplatin. The disulfide-rich scaffold was rationally designed to block cisplatin depletion by GSH in cancer cells, thereby circumventing the chemoresistance of cisplatin. Besides, the switch NPs-cisplatin were designed with an acidic stimulus-responsiveness, being capable of masking and preventing cisplatin depletion by other macromolecules in human body but being a switch-on mode just in the cancer cells. By the rational design, engineering, mechanism elucidation, and functional verification in vitro and in vivo, we aim at offering a kind of disulfide switchable nanoparticles complexing cisplatin for circumventing chemoresistance of breast cancer.
Experimental methods include design and synthesis of disulfide switchable nanoparticles complexing cisplatin, characterization of switch NPs-cisplatin, cellular uptake and the mechanism in the resistant breast cancer cells, cytotoxicity and apoptosis in breast cancer cells and anticancer efficacy in the resistant breast cancer-bearing mice. See experimental details in Supporting information.
Switch NPs-cisplatin were rationally designed by linking HA with Pasp for synthesizing the disulfide-rich polymer (HA-ss-Pasp), which was further complexed with cisplatin, and prepared into nanoparticles by self-assembly (Fig. 1A and Fig. S1 in Supporting information). At first of all, hyaluronic acid (HA) was reduced by sodium cyanoborohydride to obtain the open-loop HA. Secondly, open-loop HA was reacted with cystamine hydrochloride to form a hyaluronic acid conjugate (HA-ss). Thirdly, HA-ss was further linked with Pasp using EDC (1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride) and NHS (N-hydroxysuccinimide) by amidation reaction to form HA-ss-Pasp (Figs. S2A and B in Supporting information). Finally, HA-ss-Pasp was conjugated with cisplatin through complexation to obtain the end product of switch NPs-cisplatin.

|
Download:
|
| Fig. 1. Design and synthesis of disulfide switchable nanoparticles complexing cisplatin. (A) Synthesis route. (B) TEM analyses. Transmission electron microscope (TEM) images of switch NPs-cisplatin in different conditions: (i) PBS pH 7.4; (ii) after incubated in 10 mmol/L GSH for 24 h; (iii) after incubated in PBS (pH 5.5) for 24 h. The results indicate that the switch NPs-cisplatin has pH responsiveness, and GSH responsiveness. | |
{kind=link}
To verify the conjugation between cisplatin and Pasp, the study was performed by thermal gravimetric analysis. The results showed that, when temperature was below 300 ℃, the water loss curve of Pasp (Fig. S2C(i) in Supporting information) was declined more rapidly, as compared to that of Pasp-cisplatin (Fig. S2C(ii) in Supporting information), indicating a successful complexation of Pasp-cisplatin.
To characterize the structure of switch NPs-cisplatin, the studies were performed by infrared (IR) spectroscopy and 1H nuclear magnetic resonance (NMR) spectroscopy, respectively. The result from IR study showed that the characteristic peaks were at 1715.00 cm-1 for HA (red shadow), and 1000.00–1200.00 for Pasp (blue shadow), indicating a successful synthesis of HA-ss-Pasp (Fig. S2A). Besides, the results from NMR study showed that the characteristic peaks were at 3.0–4.0 ppm, around 2 ppm for HA (red shadow), and around 2.5 ppm for Pasp (green shadow), further demonstrating the successful synthesis of HA-ss-Pasp (Fig. S2B).
Cisplatin is a cell cycle non-specific agent that disrupts DNA function and inhibits cell mitosis [31,32]. It has a wide antitumor spectrum and is widely used in digestive tract tumors [33], respiratory tract tumors [34], gynecological tumors [35], and head-neck malignant tumors [36]. Its prominent toxic and adverse reactions in the clinical uses have been evidenced with renal toxicity [37,38] that is mainly manifested as transient renal damage, and with digestive tract adverse reactions [39] that are manifested as nausea, vomiting, diarrhea and other digestive tract reactions. Furthermore, cisplatin demonstrates strong drug resistance, which is caused by the interaction of multiple resistant factors, one of which is the cisplatin depletion by GSH, resulting in the loss of efficacy of cisplatin. Moreover, GSH has a higher concentration in the resistant cancer cells [40], further undermining the efficacy of cisplatin. Consequently, we design the disulfide switchable nanoparticles complexing cisplatin (switch NPs-cisplatin) that aim at blocking the cisplatin depletion by GSH, in addition to covering the direct exposure of cisplatin in normal tissue, and diminishing the toxic and adverse reactions in the treatment of breast cancer.
In the rational design, Pasp is selected as the biocompatible scaffold material because its side chain group carboxyl is able to react with many drugs, such as cisplatin [41,42], hydroxycamptothecin [43], and to be easily modified on polymer blocks [44,45]. In the study, mild condition (ambient temperature) and simple synthetic methods (redox and amidation reactions) are optimized and applied for the potential industrial production. By linking Pasp with a disulfide bond to hyaluronic acid (HA) (Fig. 1, Figs. S1 and S2), the scaffold material is self-assembled into nanoparticles by the preparation processing.
To optimize and characterize the switch NPs-cisplatin, the particle size and zeta potential were evaluated by dynamic light scattering (DLS) method, and the encapsulation efficiency was measured by ultraviolet spectroscopy. The results from the formulation screening showed the particle size and zeta potential values were changed with different feeding ratios (Table S1 in Supporting information). Based on the screening, the ratio of Pasp to HA was optimized at a value of 1:1, and the particle size, polydispersity index, zeta potential and encapsulation efficiency for the switch NPs-cisplatin were 152.9 ± 5.5 nm, 0.267 ± 0.007, −11.2 ± 0.5 mV, and 93.4%, respectively (Fig. S3A and Table S2 in Supporting information).
To understand the critical micelle concentration (CMC) of switch NPs-cisplatin, the study was performed by ultraviolet spectroscopy using pyrene as a probe. The results showed that, with the increase of switch NPs-cisplatin concentration (C) in the aqueous system, the profile of pyrene absorbance ratio versus lgC exhibited a linear rise, while occurred an inflection point at a certain concentration, followed by a precipitous decline, indicating that the formation of switch NPs-cisplatin happened at 0.01 mg/mL CMC (Fig. S3B in Supporting information).
To study the drug release of switch NPs-cisplatin, the studies were performed in pH 7.4 PBS and pH 5.5 PBS by ultraviolet spectroscopy, respectively. The results showed that the release profile exhibited an immediate rise at 0.5 h (up to 80% cumulative release of cisplatin at 20 h) in the pH 5.5 PBS but a minimal increase, followed by a flat line (below 10% cumulative release of cisplatin at 20 h) in the pH 7.4 PBS (Fig. S3C in Supporting information).
To study the pH responsiveness and GSH responsiveness of switch NPs-cisplatin, the studies were performed by DLS and TEM, respectively. The results from DLS measurements showed that the particle size was increased by 2.5 times in pH 5.5 PBS solution, increased by 5.7 times in the pH 7.4 PBS containing 10 mmol/L GSH, and increased by 6.6 times in pH 5.5 PBS solution containing 10 mmol/L GSH, as compared to that in pH 7.4 PBS solution (Figs. S3D(ⅰ) and (ⅱ) in Supporting information). The results from TEM observations results showed that the particles exhibited the intact spherical nanoparticles in pH 7.4 PBS solution, but the dissociated particle fragments in pH 7.4 PBS solution containing GSH (10 mmol/L), and the loose foggy particles in pH 5.5 PBS (Fig. 1B).
The obtained nanoparticles are stable in the normal body fluid environment (about pH 7.4), while they become unconsolidated in the acidic tumor masses (pH 5.5), and are cleavable in the cancer cells (by rich GSH, approximately 40 mmol/L [15]) (Fig. 1B, Tables S1 and S2).
To investigate the cellular uptake effect, the studies were performed on the drug-resistant breast cancer cells (MCF-7/adr) by flow cytometry (FCM). The result from FCM showed that the uptake effect of fluorescence-labeled switch NPs Cou was significantly higher (up to 2 folds) than free coumarin (Fig. S4A in Supporting information).
To further observe the cellular uptake effect, the studies were performed on the resistant MCF-7/adr cells by confocal laser scanning microscopy (CLSM). The results showed that, under a direct observation, the fluorescence intensity of switch NPs Cou uptake was evidently stronger than that of free coumarin (Fig. S4B(i) in Supporting information). The results from 2.5D diagram and fluorescence quantification analyses by ImageJ showed that the cellular uptake of switch NPs Cou was 3 folds higher than that of free coumarin (Fig. S4B(ii) in Supporting information). To reveal the uptake mechanism, varying formulations were employed for comparisons by FCM, including switch NPs Cou plus colchicine (macropinocytosis inhibitor), switch NPs Cou plus methyl-β-cyclodextrin (MβCD, caveolin/lipid raft-mediated endocytosis inhibitor), switch NPs Cou plus quercetin (non-clathrin and non-caveolin inhibitor), and switch NPs Cou plus 4 ℃ condition (ATP energy inhibitor). The results showed that low temperature (4 ℃), MβCD, quercetin, and colchicine could significantly decrease the cellular uptake of switch NPs Cou by over 90%, by 70%, by 30%, and by 20%, as compared to that of switch NPs Cou alone, respectively (Fig. S4C in Supporting information).
The results from the cellular uptake demonstrate that the switch NPs exhibit the strongest uptake effect, indicating an evident targeting effect on the resistant breast cancer cells. It has been reported that the disaccharide unit of hyaluronic acid can specifically bind with the CD44 receptor on the surface of cancer cells [46], and the resistant breast cancer cells have been identified as highly expressed CD44 receptor [47]. Accordingly, the hyaluronic acid coating layer on the surface of switch NPs-cisplatin provides the premise for the targeting effect. Meanwhile, the uptake mechanism study reveal that the switch NPs could be internalized by the resistant cancer cells through caveolin/lipid raft-mediated active transport [48], in which ATP provides the power for the endocytosis.
To verify the GSH depletion by disulfide-rich scaffold (HA-ss-Pasp) in the GSH solution, the changes of GSH concentration were measured by GSH detection kit. To investigate the GSH depletion by HA-ss-Pasp in the resistant MCF-7/adr cells, the changes of GSH concentration were further measured by GSH detection kit, using buthionine-sulfoximine (BSO) as a positive control. The results showed that both BSO and disulfide-rich HA-ss-Pasp significantly decreased the GSH level of the resistant cancer cells, as compared to the blank control (Fig. S4D in Supporting information).
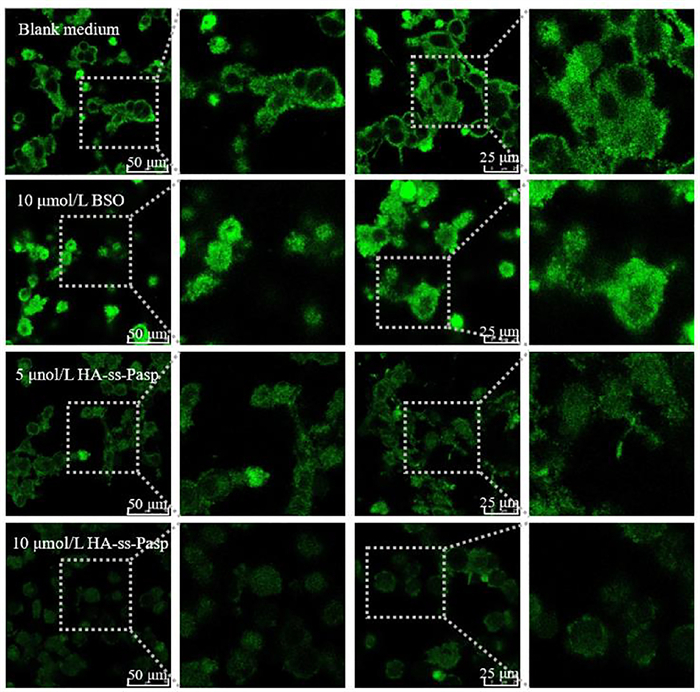
To further directly observe the GSH depletion by HA-ss-Pasp, the studies were performed on the resistant MCF-7/adr cells by viewing GSH fluorescence intensity using CLSM. The results showed that the GSH depletion by HA-ss-Pasp in the resistant cancer cells could be evidently observed, in viewing the decrease of the fluorescence intensity (10 µmol/L BSO > 5 µmol/L HA-ss-Pasp > 10 µmol/L HA-ss-Pasp) (Fig. 2). The results from 2.5D diagram and quantification with the software ImageJ showed that the GSH levels were decreased 1.3 folds by BSO, and 4.7 folds by HA-ss-Pasp, as compared to that by blank control (Fig. S4E in Supporting information), confirming a significant GSH depletion effect.

|
Download:
|
| Fig. 2. Confocal laser scanning microscopy observation on GSH depletion. The results from the cellular uptake demonstrate that the switch NPs exhibit the strongest uptake effect, and disulfide-rich scaffold of switch NPs-cisplatin exerts a stronger effect than the positive control in viewing the GSH depletion status. | |
{kind=link}
The GSH depletion by switch NPs is designed for blocking the cisplatin depletion by GSH in the resistant cancer cells. The results reveal that the disulfide-rich scaffold material is able to reach this purpose, namely, to reduce the inactivation of cisplatin due to its complexing with GSH in the resistant breast cancer cells. In the study, buthionine-sulfoximine (BSO) is included as a positive control due to the fact that it is a GSH synthase inhibitor, which results in the decrease of GSH in the cells by preventing the synthesis of intermediate dipeptide from glutamic acid of GSH [49]. The results indicate that the disulfide-rich scaffold of switch NPs-cisplatin exerts a stronger effect than the positive control in viewing the GSH depletion status.
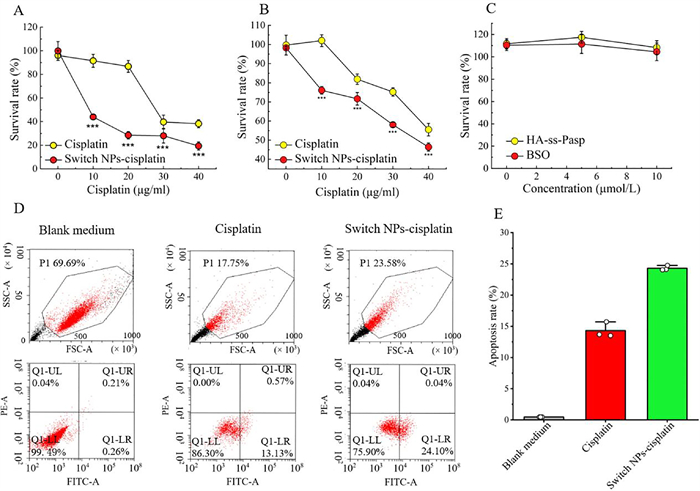
To evaluate the cytotoxicity of switch NPs-cisplatin in the resistant MCF-7/adr cells, the studies were performed by cell counting kit-8 (CCK8) kit. The results showed the switch NPs-cisplatin exhibited significantly stronger killing effect than free cisplatin in the resistant MCF-7/adr cells, ether in serum-free or in serum-containing culture medium (Figs. 3A and B). The killing effect of switch NPs-cisplatin displayed stronger cytotoxicity than free cisplatin in the resistant MCF-7/adr cells in serum-containing culture medium due to the fact that part of free cisplatin was depleted by the serum components. The results from the half inhibitory concentration (IC50) measurements showed that the IC50 values were 2.47 µg/mL for switch NPs-cisplatin, and 9.91 µg/mL for free cisplatin in the resistant MCF-7/adr cells in the serum-free culture medium. Besides, the IC50 values were 1.64 µg/mL for free cisplatin in the non-resistant MCF-7 cells in the serum-free culture medium, indicating that the drug resistance was increased by 6 folds (resistance index, 6.04) in the MCF-7/adr cells (Fig. S5 in Supporting information). Results from the evaluation on the controls showed that both BSO and blank HA-ss-Pasp did not affect the survival of the cancer cells (Fig. 3C).

|
Download:
|
| Fig. 3. Cytotoxicity and apoptosis in the resistant breast cancer cells. (A) Killing effect in the resistant breast cancer cells in serum-free culture medium. (B) Killing effect in the resistant breast cancer cells in serum-containing culture medium. (C) Cytotoxic evaluations of HA-ss-Pasp and BSO in the resistant breast cancer cells. (D, E) Inducing apoptosis of the resistant breast cancer cells. (D) Dot plots for the induced apoptosis. (E) Quantification for the induced apoptosis. | |
{kind=link}
To evaluate the inducing apoptosis by switch NPs-cisplatin in the resistant MCF-7/adr cells, the studies were performed by FCM. The results for FCM diagram showed that the switch NPs-cisplatin evidently enhanced apoptosis of the resistant cancer cells, as compared to free cisplatin (Fig. 3D), and that the apoptosis rates induced by switch NPs-cisplatin and by free cisplatin were 24.31% ± 0.38%, and 14.31% ± 1.22%, respectively (Fig. 3E). The results from microscopy showed that the switch NPs-cisplatin resulted in more severe damages of the resistant MCF-7/adr cells, exhibiting partially shrunk shape (in apoptotic form) and mostly swollen round shape (in necrotic form) (Fig. S6 in Supporting information).
The overall killing effect of chemotherapeutic agent could be reflected by the cytotoxicity that may derive from both the direct necrosis by DNA damage and the apoptosis by the programmed suicide of the cancer cells [50]. The results from cytotoxicity demonstrate that the switch NPs-cisplatin have stronger killing effect than free cisplatin in the resistant breast cancer cells, confirming that the smart nanoparticles could effectively enhance the therapeutic effect, and be able to overcome the chemoresistance of cisplatin. The results from apoptosis and cell morphology observation further verify that, in addition to the direct necrosis by DNA damage, the switch NPs-cisplatin also initiate more strong apoptosis of the resistant breast cancer cells, then potentiating the therapeutic effect.
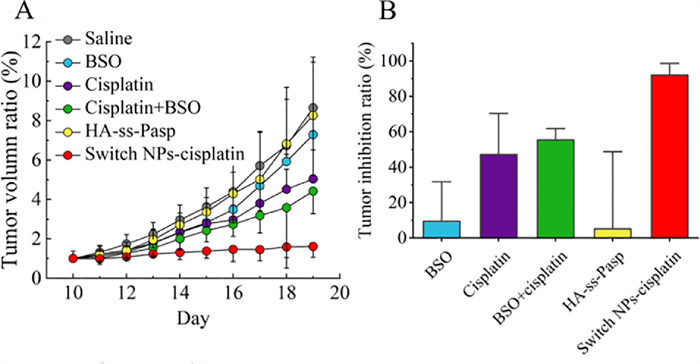
To evaluate the anticancer efficacy of switch NPs-cisplatin, the MCF-7/adr cells were inoculated into Balb/c mice for establishing the resistant breast cancer-bearing mice (No. LA2021252), and varying formulations were administrated at day 12, 15 and 18 by intravenous injection via tail vein (physiological saline, free cisplatin, HA-ss-Pasp and switch NPs-cisplatin), and by peritumoral injection (BSO), respectively (Fig. S7A in Supporting information).The results showed that the switch NPs-cisplatin could nearly fully inhibit the growth of tumor masses (Fig. 4A), and that the tumor inhibition ratios were 92% for switch NP-cipalin, 47% for free cisplatin, 55% for free cisplatin plus BSO, 9% for BSO, and 5% for HA-ss-Pasp, respectively (Fig. 4B).

|
Download:
|
| Fig. 4. Anticancer efficacy in the resistant breast cancer-bearing mice. (A) Tumor volume. (B) Tumor inhibition ratio. The results indicate that the switch NPs-cisplatin remarkably enhance the anticancer efficacy as compared to free cisplatin. In addition, the switch NPs-cisplatin are able to ameliorate the liver fibrosis and pulmonary congestion of animals as compared to free cisplatin. | |
{kind=link}
To preliminary evaluate the formulation safety, the body weight changes of the resistant breast cancer-bearing mice were monitored once daily since the day of drug administration. The results showed that all formulations did not significantly affect the body weight, except for free cisplatin that resulted in significant body weight loss (Fig. S7B in Supporting information).
Besides, the blood routine indicators were also analyzed. The results showed that the formulations did not influence the indicators, except for free cisplatin, and free cisplatin plus BSO which caused an increase of neutrophils while a decrease of lymphocytes in the resistant cancer-bearing mice (Table S3 in Supporting information). To further understand the effects of switch NPs-cisplatin on the organs, the major organ tissues (heart, liver, spleen, lung and kidney) were isolated from the resistant cancer-bearing mice after sacrificing the animals, and studied by the hematoxylin-eosin (HE) staining analysis. The results showed that the switch NPs-cisplatin did not evidently affect the organ tissues. In contrast, free cisplatin, and cisplatin plus BSO caused pulmonary congestion, and liver fibrosis (Fig. S7C in Supporting information).
The anticancer efficacy and the preliminary safety are further evaluated on the resistant breast cancer-bearing mice, and the results demonstrate that, either the efficacy or the safety concern, the switch NPs-cisplatin exhibit superior strengths to free cisplatin for the potential clinical application.
In summary, a novel type of smart disulfide switchable nanoparticles complexing cisplatin (switch NPs-cisplatin) was rationally designed, and engineered by synthesizing a hyaluronic acid disulfide bonded polyaspartic acid (HA-ss-Pasp) and complexing cisplatin. The results showed that the switch NPs-cisplatin had a nanoscale of particle size (150 nm), higher drug encapsulation efficiency (> 90%), and suitable drug release profile. They demonstrated evident pH responsiveness and GSH responsiveness, and targeting effect in the resistant breast cancer cells. Furthermore, they were able to block the cisplatin depletion by GSH in the resistant cancer cells, thus circumventing the chemoresistance. Consequently, switch NPs-cisplatin displayed a remarkable killing effect in the resistant breast cancer cells in vitro, and in the resistant breast cancer-bearing mice. In conclusion, switch NPs-cisplatin could be used as a smart formulation of cisplatin for overcoming the chemoresistance of breast cancer. The present study also provides a universal drug delivery carrier platform for high efficient but low systemic toxic chemotherapy.
Declaration of competing interestThe authors declare no competing interests.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 81874303, 82173752).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107744.
| [1] |
G.W. Sledge, B.J. Roth, Semin. Oncol. 16 (1989) 110-115. |
| [2] |
M. Xiang, A.D. Colevas, F.C. Holsinger, Q.T.X. Le, B.M. Beadle, J. Natl. Compr. Cancer Netw. 17 (2019) 1065-1073. DOI:10.6004/jnccn.2019.7297 |
| [3] |
S. Bouberhan, E. Pujade-Lauraine, S.A. Cannistra, J. Clin. Oncol. 37 (2019) 2424-2436. DOI:10.1200/jco.19.00314 |
| [4] |
J. Guigay, A. Aupérin, J. Fayette, et al., Lancet Oncol. 22 (2021) 463-475. DOI:10.1016/S1470-2045(20)30755-5 |
| [5] |
X. Yang, M. Fraser, U.M. Moll, A. Basak, B.K. Tsang, Cancer Res. 66 (2006) 3126-3136. DOI:10.1158/0008-5472.CAN-05-0425 |
| [6] |
R. Arriagada, B. Bergman, A. Dunant, et al., N. Engl. J. Med. 350 (2004) 351-360. DOI:10.1056/NEJMoa031644 |
| [7] |
T. Qin, B. Li, X. Feng, et al., J. Exp. Clin. Cancer Res. 37 (2018) 287. DOI:10.1186/s13046-018-0934-9 |
| [8] |
L. Galluzzi, L. Senovilla, I. Vitale, et al., Oncogene 31 (2012) 1869-1883. DOI:10.1038/onc.2011.384 |
| [9] |
J. Molnár, H. Engi, J. Hohmann, et al., Curr. Top Med. Chem. 10 (2010) 1757-1768. DOI:10.2174/156802610792928103 |
| [10] |
S.Y. Loh, P. Mistry, L.R. Kelland, G. Abel, K.R. Harrap, Br. J. Cancer 66 (1992) 1109-1115. DOI:10.1038/bjc.1992.419 |
| [11] |
S.C. Shuck, E.A. Short, J.J. Turchi, Cell Res. 18 (2008) 64-72. DOI:10.1038/cr.2008.2 |
| [12] |
M. Yuan, P. Luong, C. Hudson, K. Gudmundsdottir, S. Basu, Cell Death Dis. 1 (2010) e16. DOI:10.1038/cddis.2009.15 |
| [13] |
A. Citri, Y. Yarden, Nat. Rev. Mol. Cell Biol. 7 (2006) 505-516. DOI:10.1038/nrm1962 |
| [14] |
S. Akiyama, Z.S. Chen, T. Sumizawa, T. Furukawa, Anticancer Drug Des. 14 (1999) 143-151. |
| [15] |
A. Bansal, M.C. Simon, J. Cell Biol. 217 (2018) 2291-2298. DOI:10.1083/jcb.201804161 |
| [16] |
L. Kennedy, J.K. Sandhu, M.-.E. Harper, M. Cuperlovic-Culf, Biomolecules 10 (2020) 1429. DOI:10.3390/biom10101429 |
| [17] |
C.R.R. Rocha, C.C.M. Garcia, D.B. Vieira, et al., Cell Death Dis. 5 (2014) e1505. DOI:10.1038/cddis.2014.465 |
| [18] |
A.K. Godwin, A. Meister, P.J. O'Dwyer, et al., Proc. Natl. Acad. Sci. U. S. A. 89 (1992) 3070-3074. DOI:10.1073/pnas.89.7.3070 |
| [19] |
Y. Su, Y. Liu, X. Xu, et al., ACS Appl. Mater. Interfaces 10 (2018) 38700-38714. DOI:10.1021/acsami.8b11349 |
| [20] |
A. Shikanov, S. Shikanov, B. Vaisman, J. Golenser, A.J. Domb, Chemother. Res. Pract. 2011 (2011) 175054. |
| [21] |
L. Tang, A. Zhang, Z. Zhang, et al., Cancer Commun. (Lond) 42 (2022) 141-163. DOI:10.1002/cac2.12255 |
| [22] |
J. Hu, X. Yuan, F. Wang, et al., Chin. Chem. Lett. 32 (2021) 1341-1347. DOI:10.1016/j.cclet.2020.11.006 |
| [23] |
J. Yan, Y. Zhang, L. Zheng, et al., Chin. Chem. Lett. 33 (2022) 767-772. DOI:10.1016/j.cclet.2021.08.018 |
| [24] |
P.S Yavvari, A.K Awasthi, A Sharma, A Bajaj, A Srivastava, J. Mater. Chem. B 7 (2019) 2102-2122. DOI:10.1039/C8TB02962H |
| [25] |
M. Frankel, A. Berger, Nature 163 (1949) 213. DOI:10.1038/163213b0 |
| [26] |
M.F.P. Graça, S.P. Miguel, C.S.D. Cabral, I.J. Correia, Carbohydr. Polym. 241 (2020) 116364. DOI:10.1016/j.carbpol.2020.116364 |
| [27] |
S. Vasvani, P. Kulkarni, D. Rawtani, Int. J. Biol. Macromol. 151 (2020) 1012-1029. DOI:10.1016/j.ijbiomac.2019.11.066 |
| [28] |
X.Y. Lu, D.C. Wu, Z.J. Li, G.Q. Chen, Prog. Mol. Biol. Transl. Sci. 104 (2011) 299-323. |
| [29] |
C. Elvira, A. Gallardo, J.S. Roman, A. Cifuentes, Molecules 10 (2005) 114-125. DOI:10.3390/10010114 |
| [30] |
B. Wang, S. Wang, Q. Zhang, et al., Acta Biomater. 96 (2019) 55-67. DOI:10.1016/j.actbio.2019.05.044 |
| [31] |
E. Reed, Cancer Chemother. Biol. Response Modif. 13 (1992) 83-90. |
| [32] |
F.M. Muggia, Semin. Oncol. (1991) 1-4. |
| [33] |
J. Lee, D.H. Lim, S. Kim, et al., J. Clin. Oncol. 30 (2012) 268-273. |
| [34] |
L. Fournel, Z. Wu, N. Stadler, et al., Cancer Lett. 464 (2019) 5-14. DOI:10.1016/j.canlet.2019.08.005 |
| [35] |
K.K. Crean-Tate, C. Braley, G. Dey, et al., J. Ovarian Res. 14 (2021) 55. DOI:10.1186/s13048-021-00797-x |
| [36] |
J. Bernier, J.S. Cooper, T.F. Pajak, et al., Head Neck 27 (2005) 843-850. DOI:10.1002/hed.20279 |
| [37] |
J. Zhang, Z.-.W. Ye, K.D. Tew, D.M. Townsend, Adv. Cancer Res. 152 (2021) 305-327. DOI:10.3390/mi12030305 |
| [38] |
D. Sheikh-Hamad, K. Timmins, Z. Jalali, J. Am. Soc. Nephrol. 8 (1997) 1640-1644. |
| [39] |
M. Chovanec, M. Abu Zaid, N. Hanna, et al., Ann. Oncol. 28 (2017) 2670-2679. DOI:10.1093/annonc/mdx360 |
| [40] |
N. Traverso, R. Ricciarelli, M. Nitti, et al., Oxid. Med. Cell Longev. 2013 (2013) 972913. |
| [41] |
P. Zhang, K. Yuan, C. Li, et al., Macromol. Biosci. 17 (2017) 1700206. DOI:10.1002/mabi.201700206 |
| [42] |
X. Yao, C. Xie, W. Chen, et al., Biomacromolecules 16 (2015) 2059-2071. DOI:10.1021/acs.biomac.5b00479 |
| [43] |
G.B. Ding, Y. Wang, Y. Guo, L. Xu, ACS Appl. Mater. Interfaces 6 (2014) 16643-16652. DOI:10.1021/am503359g |
| [44] |
H. Adelnia, I. Blakey, P.J. Little, H.T. Ta, Front. Chem. 7 (2019) 755. DOI:10.3389/fchem.2019.00755 |
| [45] |
A. Fontana-Escartín, G. Ruano, F.M. Silva, et al., Int. J. Mol. Sci. 22 (2021) 13165. DOI:10.3390/ijms222313165 |
| [46] |
I. Morath, T.N. Hartmann, V. Orian-Rousseau, Int. J. Biochem. Cell Biol. 81 (2016) 166-173. DOI:10.1016/j.biocel.2016.09.009 |
| [47] |
C. Yang, Y. Sheng, X. Shi, et al., Cell Death Dis. 11 (2020) 831. DOI:10.1038/s41419-020-03037-0 |
| [48] |
L.A. Mulcahy, R.C. Pink, D.R.F. Carter, J. Extracell Vesicles 3 (2014) 24641. |
| [49] |
R. Drew, J.O. Miners, Biochem. Pharmacol. 33 (1984) 2989-2994. DOI:10.1016/0006-2952(84)90598-7 |
| [50] |
E. Pérez-Herrero, A. Fernández-Medarde, Eur. J. Pharm. Biopharm. 93 (2015) 52-79. DOI:10.1016/j.ejpb.2015.03.018 |