 2023, Vol. 34
2023, Vol. 34 


b State Key Laboratory of Solidification Processing and School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, China
Owing to their lower zero-point energy, the chemical stability of C–D bonds is considerably superior to C–H bonds [1]. Accordingly, deuterated molecules and polymers have been extensively utilized as valuable standards in analytical chemistry, probes for metabolism and pharmacokinetics [2,3], labeling tools for elucidating reaction mechanisms [4], special materials like deuterated polymers for laser inertial confinement fusion (ICF) [5], etc. As useful building blocks or end products, the synthesis of deuterated terminal alkenes (e.g., styrene) have been widely reported [6-8]. By contrast, deuterated ethylene is a key commodity chemical for synthesizing various deuterated products including deuterated polyolefins (polyethylene, etc.) and deuterated chemicals (e.g., ethylene oxide, acetic acid, acetaldehyde), but its low-cost and large-scale production still remain a grand challenge.
The H/D exchange and reductive deuteration are two universal approaches for selective incorporation of deuterium into vinyl groups or alkenes [9,10]. Unfortunately, for the H/D exchange, precious metal-based catalysts and harsh reaction conditions (strong acid/base or high temperature) are essential for promoting the substitution of deuterium atoms for hydrogen atoms [11-13]. The H/D exchange also suffers from low deuteration efficiency, poor selectivity of deuterated sites and numbers [14]. Considering that alkenes are commonly synthesized by selective semi-hydrogenation of the corresponding alkynes, reductive deuteration of alkynes is a straightforward strategy for synthesizing target deuterated alkenes. Nevertheless, reductive deuteration of alkynes often requires expensive and unrecoverable D2 [15] or other deuterium reagents (ethanol-d6 [16], EtOD-d1 [17], NaOD-D2O [18], etc.) as deuterium source and noble metal Pd-based catalysts for activating reactants at relatively high reaction temperature. Therefore, the exploration of a high-efficiency reductive deuteration strategy of alkynes with cheap and safe deuterium oxide (D2O) as deuterium source is greatly appealing. Owing to its environmental benignity and ambient reaction conditions, electrochemically reductive deuteration of alkynes is such a promising approach for producing deuterated alkenes. For that, Zhang [19] and Berlinguette's [20] groups successively reported the electrochemically reductive deuteration of alkynes including long-chain terminal alkynes and phenylacetylene-derived alkynes. However, the utilization of organic solvents and low production rate of deuterated alkenes seriously limit their large-scale implementations. Nowadays, the electrocatalytic reductive deuteration of acetylene to synthesize commodity deuterate ethylene still remains unexplored due to poor dissolution and diffusion of acetylene in water or organic solvents.
In this work, we first highlight a novel electrocatalytic acetylene deuteration strategy for producing deuterated ethylene on Ag nanoparticles (Ag NPs) under ambient conditions, where deuterium oxide serves as the D source. Unprecedentedly, in alkaline D2O solution, the deuterated ethylene Faradaic efficiency (FE) and production rate of Ag NPs reach 99.3% at –0.6 V vs. reversible hydrogen electrode (RHE) and 3.72 × 103 mmol h−1 gcat−1 in the flow cell, respectively, which far exceed 1.87 × 102 mmol h−1 gcat−1 for previously reported deuterated alkenes. Meanwhile, the deuterated ethylene FE of Ag NPs was steadily larger than 95% over a 40 h stability measurement in a large two-electrode flow cell. Mass spectrometry qualitatively confirms the synthesis of semi-deuterated and perdeuterated ethylene products over electrocatalytic deuteration process. In-situ electrochemical Infrared absorption (IRAS) and Raman spectroscopic investigations reveal excellent acetylene adsorption and the formation of deuterated ethylene on Ag NPs.
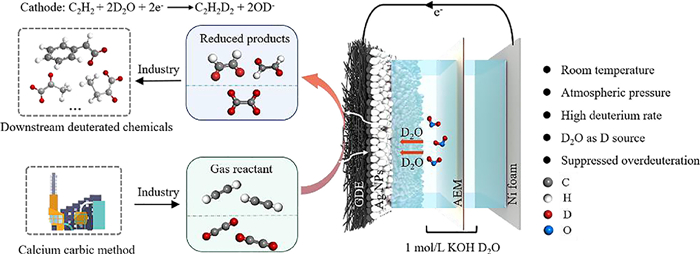
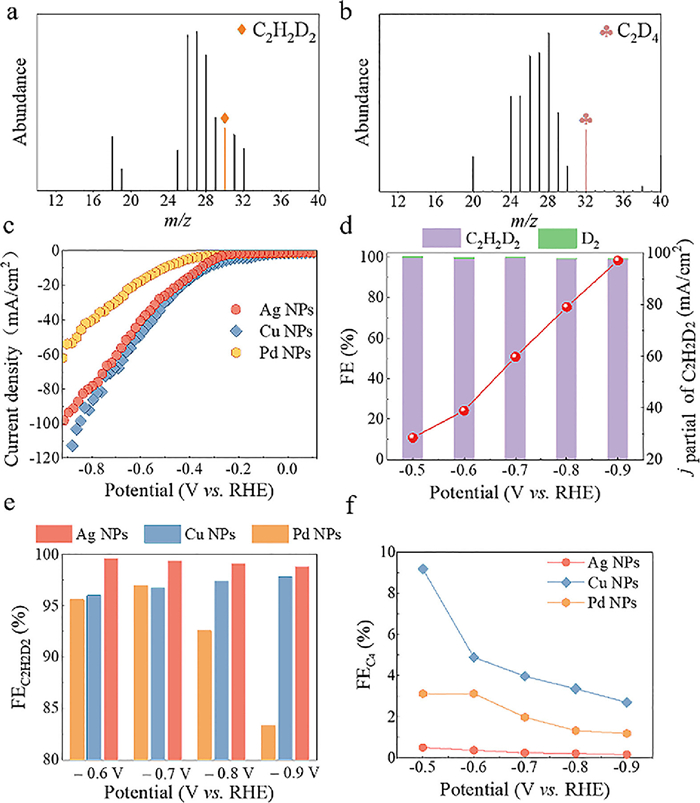
According to our previous work on electrocatalytic acetylene semi-hydrogenation [21,22], Pd nanoparticles (NPs), Cu NPs and Ag NPs were employed as model electrocatalysts for acetylene deuteration. The morphologies and chemical structures of Pd NPs, Cu NPs and Ag NPs were confirmed by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) (Figs. S1-S3 in Supporting information). The electrocatalytic acetylene deuteration was then investigated in a three-electrode flow cell (1 cm2) (Fig. S4 in Supporting information). The electrocatalysts deposited on gas diffusion electrode (GDE), nickel foam, and Hg/HgO electrode were employed as the working, counter, and reference electrodes, respectively. As shown in Fig. 1, an anion exchange membrane (AEM) separates the anodic and cathodic chambers, both of which are filled with 1 mol/L KOH D2O solution. The cathode was then fed with pure acetylene at a flow rate of 20 sccm. The polarization curves of Pd NPs under acetylene flow presented an obviously positive shift of ~320 mV in comparison with that in Ar stream (Fig. S5 in Supporting information). To investigate the possibility of electrocatalytic deuteration of acetylene to deuterated ethylene (Scheme S1 in Supporting information), the gas products on Pd NPs were qualitatively analyzed by using mass spectrometry. Fig. 2a displays a characteristic peak at m/z 30, which is attributed to semi-deuterated ethylene (C2H2D2). By contrast, no signal was recorded at m/z 30 over electrocatalytic reduction of acetylene to ethylene in 1 mol/L KOH H2O solution (Figs. S6 and S7 in Supporting information). Furthermore, after electrocatalytic deuteration of semi-deuterated acetylene, perdeuterated ethylene (C2D4) was also synthesized, which corresponded to the characteristic peak at m/z 32 in mass spectra (Fig. 2b). To exclude the disturbance of oxygen, the curve changes at m/z 32 were recorded at different gas atmosphere including Argon, perdeuterated ethylene and air (Fig. S8 in Supporting information). These results undoubtedly validate the feasibility of the electrocatalytic deuteration of acetylene to deuterated ethylene using D2O.

|
Download:
|
| Fig. 1. Schematic illustrations for electrocatalytic deuteration of acetylene to deuterated ethylene. | |
{kind=link}

|
Download:
|
| Fig. 2. The mass spectrums of semi-deuterated (a) and perdeuterated ethylene (b) on Pd NPs. (c) Polarization curves of Ag NPs, Cu NPs and Pd NPs in 1 mol/L KOH D2O solution. (d) Corresponding FE distributions of Ag NPs at –0.5 V to –0.9 V. (e) FEC2H2D2 of Ag NPs, Cu NPs and Pd NPs at different potentials. (f) The FEC4 of Ag NPs, Cu NPs and Pd NPs. | |
{kind=link}
Next, the electrocatalytic performance of Pd NPs, Cu NPs and Ag NPs were evaluated. All potentials were referred to the RHE. The FE distributions of Pd NPs during electrochemical acetylene semi-deuteration are shown in Fig. S9a (Supporting information). Clearly, the FEs of semi-deuterated ethylene (FEC2H2D2) on Pd NPs were always > 90% over applied potentials from –0.5 V to –0.8 V. However, as a result of strong competition of hydrogen evolution reaction (HER), the FEC2H2D2 of Pd NPs drastically decreased to 83% and the FED2 reached 16% at –0.9 V. The partial current density of deuterated ethylene (jC2H2D2) was 42 mA/cm2 at –0.9 V. By comparison, the FEC2H2D2 and jC2H2D2 of Cu NPs were 97% at –0.8 V and 110 mA/cm2 at –0.9 V, respectively, which were extremely higher than those for Pd NPs (Figs. S9b-d in Supporting information). Unfortunately, the FEs of deuterated 1,3-butadiene (FEC4) byproduct on Cu NPs were always larger than 3% over all applied potentials. Particularly, the FEC4 reached 9% at –0.5 V, which brought about a new impurity for semi-deuterated ethylene.
In comparison with Pd NPs and Cu NPs, Ag NPs exhibited a jC2H2D2 of 100 mA/cm2 at –0.9 V, which was much higher than 54 mA/cm2 for Pd NPs, but slightly lower than 110 mA/cm2 for Cu NPs (Fig. 2c). Fig. 2d and Fig. S10 (Supporting information) describe the FEC2H2D2 distributions and jC2H2D2 of Ag NPs. Noticeably, Ag NPs presented FEC2H2D2 of ≥98% over all applied potentials from –0.5 V to –0.9 V (Fig. 2e and Fig. S11 in Supporting information). Meanwhile, the FEC4 was only 0.2% at –0.8 V, which was far lower than 3.4% for Cu NPs and 1.3% for Pd NPs (Fig. 2f). The electrochemical double-layer capacitances (Cdl) of electrocatalysts were then estimated based on CV scans at different scan rates. As illustrated in Fig. S12 (Supporting information), the Pd NPs has a Cdl of 44.38 mF, which is lower than 56.5 mF for Ag NPs and 60.87 mF for Cu NPs. The Tafel slope of electrocatalysts is another crucial parameter for deeply probing electrocatalytic kinetics. As depicted in Fig. S13 (Supporting information), the Ag NPs presents a substantially declined Tafel slope of 189 mV/dec, which was considerably lower than 239 mV/dec for Pd NPs and 319 mV/dec for Cu NPs, suggesting an accelerated reaction kinetics for acetylene semi-deuteration. Therefore, for electrocatalytic acetylene semi-deuteration, the Ag NPs unambiguously manifested the excellent activity and selectivity. Moreover, the incorporation of H atoms in KOH into deuterated ethylene was negligible (Fig. S14 in Supporting information).
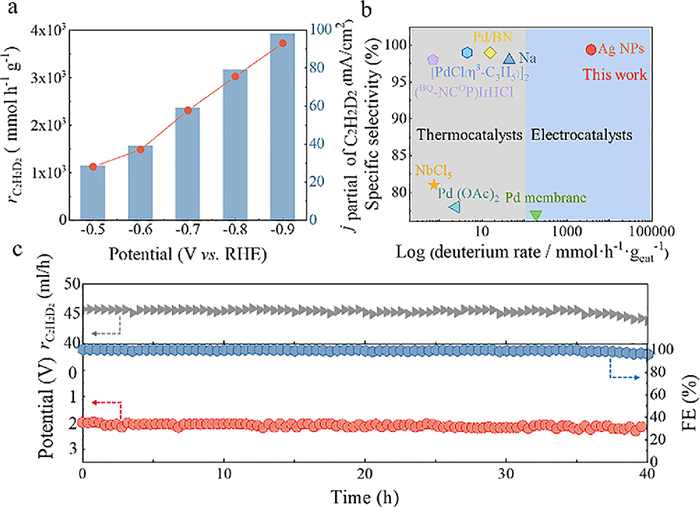
For assessing its practical implementations, the deuterated ethylene production rate (rC2H2D2) of Ag NPs was further investigated in a large two-electrode flow cell (25 cm2) by coupling NiFe-layered double hydroxide (NiFe-LDH) on nickel foam as the anode [23]. Remarkably, high FEC2H2D2 of > 94% were achieved over the cell voltages from 1.8 V to 2.8 V. Especially, negligible deuterated 1,3-butadiene was detected at 2.8 V (Fig. S15 in Supporting information). At –0.9 V in Fig. 3a, the Ag NPs perform an unprecedented rC2H2D2 of up to 3.72 × 103 mmol h−1 gcat−1 in a three-electrode flow cell, which greatly exceeds those for previously reported alkyne deuteration (Fig. 3b and Table S1 in Supporting information), e.g., 1.87 × 102 mmol h−1 gcat−1 for deuterated styrene using palladium membrane reactor. Fig. S16 (Supporting information) depicts the rC2H2D2 and partial current (I) of Ag NPs in a large two-electrode flow cell. Next, the electrocatalytic durability of Ag NPs at an acetylene flow rate of 10 mL/min was further measured in the 25 cm2 two-electrode flow cell (Fig. 3c). Obviously, the augment of cell voltage was negligible over a 40 h operation. Notably, over the long-term stability test, Ag NPs manifested FEC2H2D2 of > 95% and a stable rC2H2D2 value of 1.44 × 103 mmol h−1 gcat−1 at 100 mA. After the long-term stability test, the morphology and structural information of Ag NPs was scrutinized using the SEM, XRD, and XPS (Figs. S17-S19 in Supporting information) and no obvious variations were observed.

|
Download:
|
| Fig. 3. Electrochemical performance of Ag NPs catalysts for acetylene semi-deuteration. (a) The production rate and partial current of deuterated ethylene at different potentials in a three-electrode flow cell. (b) Comparison of the production rate and selectivity of deuterated ethylene between Ag NPs and reported catalysts. (c) Long-term stability test at a current of 100 mA in a large two-electrode flow cell (25 cm2). | |
{kind=link}
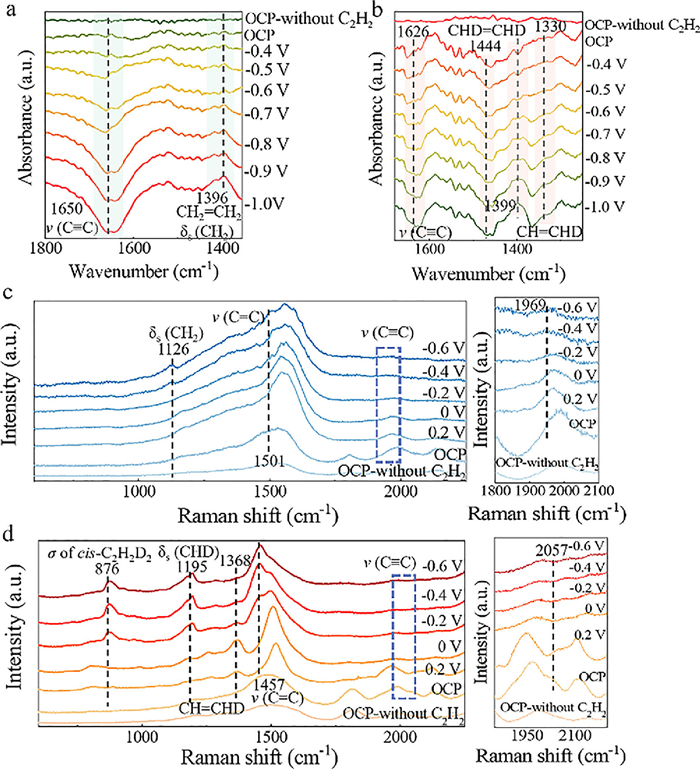
In order to profoundly reveal the kinetics of electrocatalytic acetylene deuteration on Ag NPs, the electrocatalytic acetylene hydrogenation was investigated in 1 mol/L KOH aqueous solution using a three-electrode flow cell. As indicated in Fig. S20 (Supporting information), the current density of 115 mA/cm2 for acetylene semi-hydrogenation at –0.9 V was larger than 100 mA/cm2 for acetylene semi-deuteration, which was attributed to the slower D2O dissociation kinetics [24]. Then, the in-situ electrochemical Infrared absorption and Raman spectra were further conducted for unveiling the underlying kinetics of electrocatalytic acetylene deuteration on Ag NPs. As indicated in Fig. 4a and Fig. S21a (Supporting information), the characteristic peaks of adsorbed acetylene on Ag NPs surfaces during electrocatalytic acetylene semi-hydrogenation appear at 3000-3300 cm−1 and 1650 cm−1, which correspond to ν(≡C–H) and ν(C≡C), respectively [25,26]. Along with decreased potentials from –0.4 V to –1 V, the intensities of above peaks gradually declined. Meanwhile, the distinctive signal of δs(CH2) for ethylene was detected at 1396 cm−1 from –0.7 V to –1 V, proving the semi-hydrogenation of acetylene into ethylene [27]. For electrocatalytic acetylene semi-deuteration on Ag NPs, the characteristic peaks of acetylide species (H–C≡C–H) were also observed at 3000–3300 cm−1 and 1626 cm−1 (Fig. S21b in Supporting information). Afterwards, the peaks at 1399 cm−1 and 1444 cm−1 originated from δs(CHD) and CHD-scissor modes of deuterated ethylene, respectively [28,29]. Markedly, a new peak appeared at 1330 cm−1, which might be assigned to the δ(CHD)/ν(CC) of σ-bonded CH=CHD (Fig. 4b) [25]. The peak at 1399 cm−1 might originated from ν12 of cis-configuration deuterated ethylene (cis-C2H2D2) [30].

|
Download:
|
| Fig. 4. In-situ electrochemical IR spectrum for electrocatalytic acetylene semi-hydrogenation (a) and semi-deuteration (b) reactions on Ag NPs. In-situ electrochemical Raman analyses for electrocatalytic acetylene semi-hydrogenation (c) and the semi-deuteration reactions (d) on Ag NPs. | |
{kind=link}
Next, as shown in Figs. 4c and d, for electrocatalytic acetylene semi-hydrogenation and semi-deuteration on Ag NPs, characteristic Raman peaks of v(C≡C) stretching vibration appear at 1969 cm−1 and 2057 cm−1 at open circuit potential (OCP), suggesting acetylene adsorption on the Ag NPs [31,32]. Evidently, in comparison with that for acetylene semi-hydrogenation, the vibrational frequency of C≡C bonds during acetylene showed a positive shift. Therefore, the adsorption of acetylene molecules on Ag NPs during acetylene semi-deuteration was weaker than that for acetylene semi-hydrogenation, which was well consistent with IRAS and experimental results [33]. When the potentials decreased from –0.2 V to –0.6 V, two characteristic peaks of adsorbed ethylene appeared at 1126 and 1501 cm−1, which were assigned to CH2 scissors (δs(CH2)) vibrations and v(C=C) stretch modes of π-bound ethylene, respectively (Fig. 4c) [32,34]. By contrast, the Raman peak of semi-deuterated ethylene at 1368 cm−1 was gradually observed at potentials from 0.2 V to 0 V. Along with decreased potentials, three pronounced peaks emerged at 876 cm−1, 1195 cm−1 and 1457 cm−1 (Fig. 4d), which were assigned to cis-C2H2D2 [35,36].
In conclusion, using deuterium oxide as the D source, we demonstrate the electrocatalytic semi-deuteration of acetylene to deuterated ethylene. Ag nanoparticles present an excellent electrocatalytic performance with high deuterated ethylene Faradic efficiencies, a prominent deuterium rate, and a superior long-term stability. In-situ electrochemical Raman together with Infrared absorption spectroscopy reveal the occurrence of electrocatalytic semi-deuteration of acetylene to deuterated ethylene. Therefore, this work will not only highlight a novel strategy for continuous production of deuterated ethylene, but also pave a new avenue for the efficient synthesis of other deuterated chemicals.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Natural Science Foundation of Shaanxi Province (No. 2020JQ-141), the National Natural Science Foundation of China (No. 22005245), the Synergy Innovation Foundation of the University and Enterprise for Graduate Students in Northwestern Polytechnical University (No. CX2021037), and the National Key Research and Development Program of China (No. SQ2021YFE010191). We thank the Analytical & Testing Center of Northwestern Polytechnical University for the SEM and TEM characterizations.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107765.
| [1] |
K.K. Irikura, J. Phys. Chem. Ref. Data 36 (2007) 389-397. DOI:10.1063/1.2436891 |
| [2] |
B. Belleau, J. Burba, Science 133 (1961) 102-104. DOI:10.1126/science.133.3446.102 |
| [3] |
R. Lowery, M.I. Gibson, R.L. Thompson, et al., Chem. Commun. 51 (2015) 4838-4841. DOI:10.1039/C4CC09588J |
| [4] |
E.M. Simmons, J.F. Hartwig, Angew. Chem. Int. Ed. 51 (2012) 3066-3072. DOI:10.1002/anie.201107334 |
| [5] |
M. Liu, Y. Zheng, Q. Chen, et al., J. Nucl. Mater. 535 (2020) 152-159. |
| [6] |
T.R. Puleo, A.J. Strong, J.S. Bandar, J. Am. Chem. Soc. 141 (2019) 1467-1472. DOI:10.1021/jacs.8b12874 |
| [7] |
A. Di Giuseppe, R. Castarlenas, J.J. Perez-Torrente, et al., Angew. Chem. Int. Ed. 50 (2011) 3938-3942. DOI:10.1002/anie.201007238 |
| [8] |
S.K.S. Tse, P. Xue, Z. Lin, et al., Adv. Synth. Catal. 352 (2010) 1512-1522. DOI:10.1002/adsc.201000037 |
| [9] |
E. Shirakawa, H. Otsuka, T. Hayashi, Chem. Commun. 37 (2005) 5885-5886. |
| [10] |
G. Erdogan, D.B. Grotjahn, J. Am. Chem. Soc. 131 (2009) 10354-10355. DOI:10.1021/ja903519a |
| [11] |
J. Atzrodt, V. Derdau, W.J. Kerr, et al., Angew. Chem. Int. Ed. 57 (2018) 3022-3047. DOI:10.1002/anie.201708903 |
| [12] |
M. Tinga, G. Schat, O.S. Akkerman, et al., J. Am. Chem. Soc. 115 (1993) 2808-2817. DOI:10.1021/ja00060a030 |
| [13] |
K. Harada, H. Urabe, F. Sato, Tetrahedron Lett. 36 (1995) 3203-3206. DOI:10.1016/0040-4039(95)00513-C |
| [14] |
J.C.T.E.M. Richards, R.S. Ward, D.H. Williams, J. Chem. Soc. 11 (1969) 1542-1544. |
| [15] |
Y. Yabe, Y. Sawama, Y. Monguchi, et al., Chem. Eur. J. 19 (2013) 484-488. DOI:10.1002/chem.201203337 |
| [16] |
Y. Wang, Z. Huang, X. Leng, et al., J. Am. Chem. Soc. 140 (2018) 4417-4429. DOI:10.1021/jacs.8b01038 |
| [17] |
M. Han, Y. Ding, Y. Yan, et al., Org. Lett. 20 (2018) 3010-3013. DOI:10.1021/acs.orglett.8b01036 |
| [18] |
Y. Kataoka, K. Takai, K. Oshima, et al., J. Org. Chem. 57 (1992) 1615-1618. DOI:10.1021/jo00031a057 |
| [19] |
Y. Wu, C. Liu, C. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 21170-21175. DOI:10.1002/anie.202009757 |
| [20] |
A. Kurimoto, R.S. Sherbo, Y. Cao, et al., Nat. Catal. 3 (2020) 719-726. DOI:10.1038/s41929-020-0488-z |
| [21] |
J. Bu, Z. Liu, W. Ma, et al., Nat. Catal. 4 (2021) 557-564. DOI:10.1038/s41929-021-00641-x |
| [22] |
L. Zhang, Z. Chen, Z. Liu, et al., Nat. Commun. 12 (2021) 6574. DOI:10.1038/s41467-021-26853-0 |
| [23] |
Z. Lu, W. Xu, W. Zhu, et al., Chem. Commun. 50 (2014) 6479-6482. DOI:10.1039/C4CC01625D |
| [24] |
S. Z.-Cai, Y. Shi, S. S.-Bao, et al., ACS Catal. 8 (2018) 3895-3902. DOI:10.1021/acscatal.7b04276 |
| [25] |
J. Moon, Y. Cheng, L.L. Daemen, et al., ACS Catal. 10 (2020) 5278-5287. DOI:10.1021/acscatal.0c00808 |
| [26] |
A.V. Ivanov, A.E. Koklin, E.B. Uvarova, et al., Phy. Chem. Chem. Phys. 5 (2003) 4718-4723. DOI:10.1039/b307138c |
| [27] |
J.D. Krooswyk, I. Waluyo, M. Trenary, ACS Catal. 5 (2015) 4725-4733. DOI:10.1021/acscatal.5b00942 |
| [28] |
R. Deng, J. Jones, M. Trenary, J. Phy. Chem. C 111 (2007) 1459-1466. DOI:10.1021/jp065710r |
| [29] |
L. Letendre, D.K. Liu, C.D. Pibel, et al., J. Chem. Phys. 112 (2000) 9209-9212. DOI:10.1063/1.481542 |
| [30] |
B.L. Crawford, J.E. Lancaster, R.G. Inskeep, J. Chem. Phys. 21 (1953) 678-686. DOI:10.1063/1.1698989 |
| [31] |
W.L. Parker, A.R. Siedle, R.M. Hexter, J. Am. Chem. Soc. 107 (1985) 264-266. DOI:10.1021/ja00287a055 |
| [32] |
K. Manzel, W. Schulze, M. Moskovits, Chem. Phy. Lett. 85 (1982) 183-186. DOI:10.1016/0009-2614(82)80328-X |
| [33] |
M.L. Patterson, M.J. Weaver, J. Phys. Chem. 89 (1985) 5046-5051. DOI:10.1021/j100269a032 |
| [34] |
M.F. Mrozek, M.J. Weaver, J. Phys. Chem. B 105 (2001) 8931-8937. |
| [35] |
M. de Hemptinne, J. Jungers, J. Delfrosse, Nature 140 (1937) 323-324. DOI:10.1038/140323a0 |
| [36] |
de Hemptinne C.M.M, Proc. Indian Acad. Sci. 9 (1939) 286-302. DOI:10.1007/BF03046468 |