 2023, Vol. 34
2023, Vol. 34 


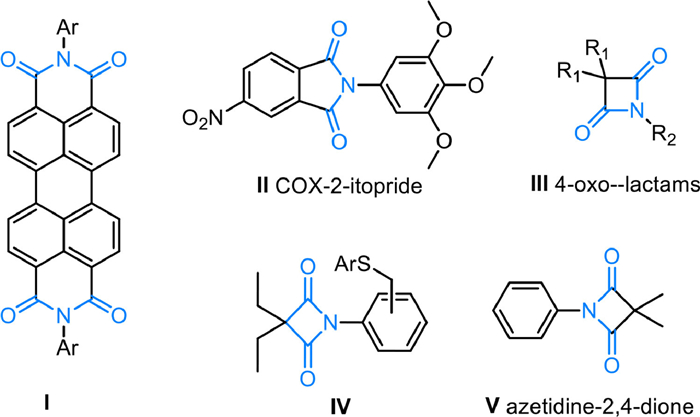
The cyclic imide scaffold as a core structural motif widely exists in a range of materials, pharmaceuticals, agrochemicals and polymer chemistry [1-5]. In particular, the amides are versatile synthetic building blocks in material science [1,6]. For example, perylenetetracarboxyl diimide Ⅰ, a brilliant charge-transport materials [7], contains an imide moiety. In addition, various important marketed drugs or prodrugs such as cycloxygenase-2-itopride with anti-inflammatory activiy Ⅱ [8], 4-oxo-lactams Ⅲ [9] with molecular recognition, Leukocyte Exfoliation inhibitors Ⅳ [10], CFRP initiators precursor Ⅴ (Fig. 1) [11], all have a cyclic imide backbone.

|
Download:
|
| Fig. 1. The representative important molecules with N-aryl cyclic imide backbone. | |
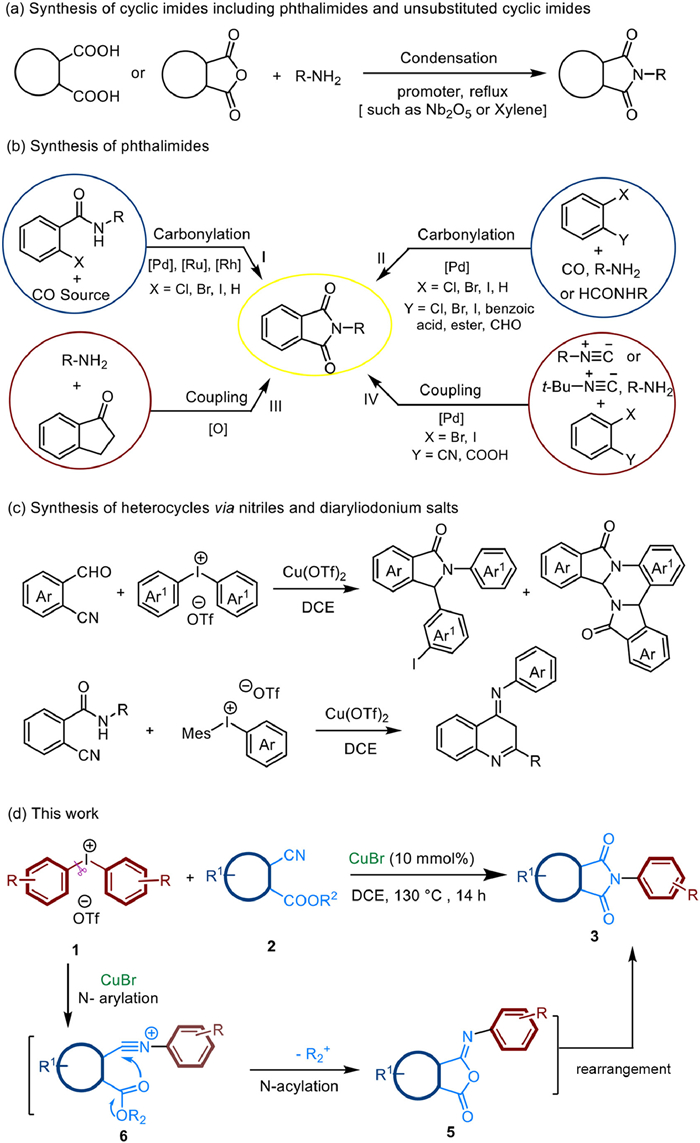
Traditionally, the cyclic imides including phthalimides and unsubstituted cyclic imides are synthesized from dicarboxylic acids or anhydrides by heating with amines (Scheme 1a) [12,13]. This method typically requires high temperatures and an excess amount of promoter (Lewis acid, base, dehydrating agent, or ionic liquids). Recent years, phthalimides have been formed via carbonylation of 1,2-diiodoarenes with amines [14-18] or benzamides with CO (Scheme 1b, Ⅰ, Ⅱ) [19-23]. Furthermore, oxidative coupling [24] and three-component coupling reactions [25,26] have expanded the range of substrates (Scheme 1b, Ⅲ, Ⅳ). In these cases, noble metals, carbonyl catalysts or CO are essential. Many efforts have been devoted to the synthesis of phthalimides. However, few effective methods for the synthesis of unsubstituted cyclic imides have been reported thus far. Herein, we would like to report a general and mild method to synthesize cyclic imides with cyanoesters and diaryliodonium salts via a tandem arylation-acylation and rearrangement sequence (Scheme 1c). This method with high tolerance of functional groups only used simple and accessible substrates. This strategy could also be applied to the fused cyclic imides such as malonimides, succinimides and glutarimides.

|
Download:
|
| Scheme 1. The methods for the synthesis of the cyclic amides. | |
During previous studies on the nitriles and diaryliodonium salts [27-39], our group developed an efficient and economical synthesis of heterocycles via the cascade reaction. Miao [40] and Novák [41] groups further applied this strategy to copper-catalyzed divergent cyclizations of 2-formylbenzomitrile and diaryliodonium salts and oxidative ring closure of ortho-cyanoanilides with hypervalent iodonium salts (Scheme 1c). Motivated by this, we envisioned that the diaryliodonium salts 1 could be attacked by cyanoesters 2 to give N-arylation intermolecular 6. Then passed through the nucleophilic addition of ester to nitriles moiety and an intramolecular 1,3-(O-N) acyl transfer rearrangenent to give target heterocycles 3 (Scheme 1d).
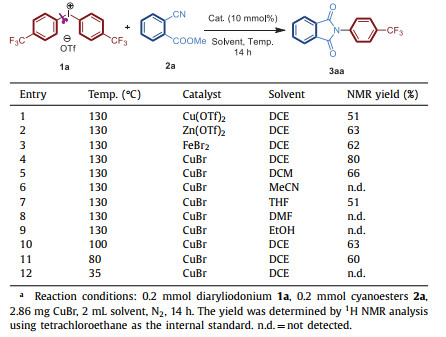
Our investigation of this Cu-catalyzed tandem process commenced with cyanoesters 2a and diaryliodonium 1a as the model to survey reaction conditions. In the presence of 10% of Cu(OTf)2 as catalyst in DCE, the N-aryl-substituted amide 3aa was detected in 51% NMR yield at 130 ℃ (Table 1, entry 1). To our delight, 3aa was obtained by changing the Cu(OTf)2 catalyst to Zn(OTf)2 or FeBr2 in 63% and 62% yields, respectively (Table 1, entries 2 and 3). Eventually, the yield was increased to 80% with CuBr as catalyst in DCE (Table 1, entry 4). The screening of solvents showed that other solvents such as DCM, THF could also enabled this transformation, but not as efficient as DCE (Table 1, entries 4, 5 and 7). However, in the presence of some highly polar or protic solvents like MeCN, DMF, EtOH, no detectable 3aa was produced (Table 1, entries 6, 8 and 9). The yield decreased at lower temperature (Table 1, entries 10-12) and no product was detected at 35 ℃.
|
|
Table 1 The optimization of synthesis of succinimide 3aa from diaryliodoni-um 1a and cyanoesters 2a in presence of catalyst.a |
{kind=link}
{kind=link}
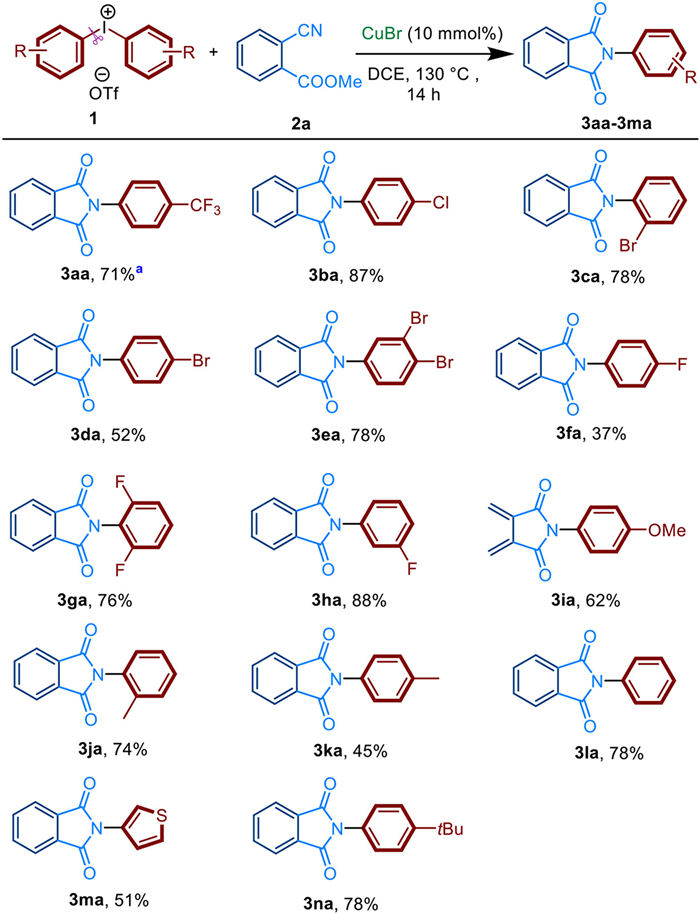
The substrate scope of the functionalized diaryliodoniums 1 was then examined (Scheme 2). Under the optimal reaction conditions, a wide range of diaryliodoniums 1 reacted with cyanoesters 2a to form N-aryl-substituted amides 3 in moderate to excellent yields. Interestingly, diaryliodoniums with a range of substituents involving halogen atoms, electron-withdrawing and electron-donating groups all worked well with 2a. Apparent substituent effect was observed. Electron-withdrawing substitutes show better effect on the reaction than electron-donating substitutes (3aa-3la). Diaryliodonium salts with electron-withdrawing groups including CF3, Cl, Br, F gave the product 3aa-3ha in 37%-88% yield. On the other hand, diaryl-iodonium salts bearing Me or MeO groups afford the corresponding imides 3ia-3ka in yields ranging from 44% to 75%. Regardless of m-substituent, o-substituent, p-substituent or di-substituent, products 3 could be obtained in moderate to good yields (up to 88%). Interestingly, S-containing heterocyclic diaryliodonium salts, which often poison noble-metal centers, was also converted into product 3ma with 51% yield. Several unsymmetric diaryliodonium salts were used to explore the preference of the transfer of aryl group in the reagents. It was found when using unsymmetric hypervalent iodine like (4-tBu-Ph-)I+MesOTf− including the aryl group with large sterically hindered group, no transfer product of Mes was detected, but only the other transfer product 3na (Scheme 2) was detected.

|
Download:
|
| Scheme 2. Substrate scope of diaryliodonium 1. Reactions were performed on 0.4 mmol scale; isolated yield. a Cyanoesters 2a was replaced by dodecyl 2-cyanobenzoate (2a'), the product 3aa was still well obtained. For 3na, unsymmetric hypervalent iodine of (4-tBu-Ph-)I+MesOTf− was used. | |
{kind=link}
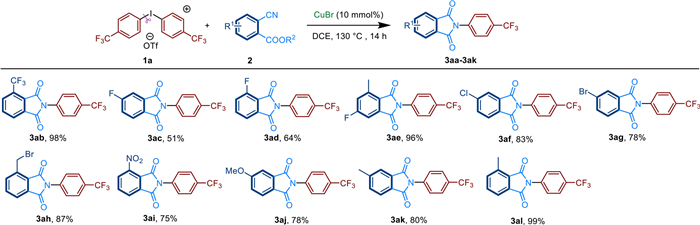
Next, the scope of the reaction with respect to aryl-cyanoesters 2 was evaluated with (4-CF3Ph)2I+OTf− 1a (Scheme 3). Electron-donating or electron-withdrawing substitutes show little effect on the reaction in moderate to good yield. Aryl-substituted cyanoesters 2, including halo (CF3, Cl, Br, F), NO2, Me and MeO substituents were all converted into the corresponding amide 3ab-3al in yields from 51% to 99%. In addition, different substituent positions, mono- or di-substituted aryl-cyanoesters 2 could react well.

|
Download:
|
| Scheme 3. Substrate scope of aryl-cyanoesters 2. Reactions were performed on 0.4 mmol scale; isolated yield. | |
{kind=link}
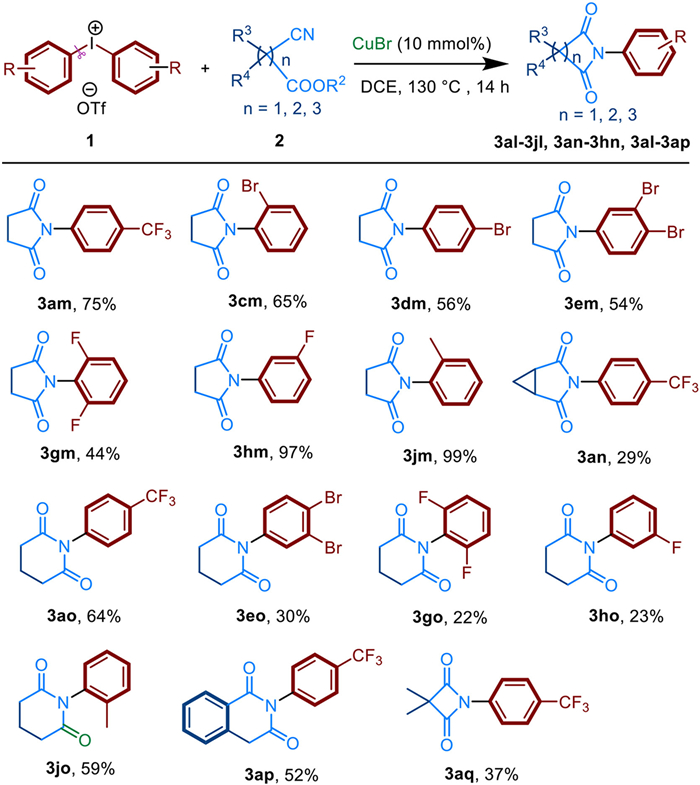
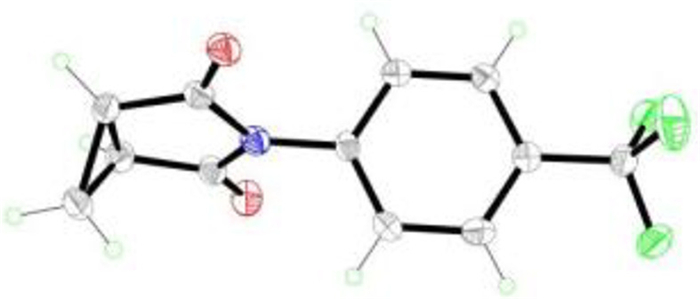
Finally, a variety of alkyl-substituted cyanoesters 2 were investigated (Scheme 4). Various unsubstituted cyclic imides, such as malonimides, succinimides and glutarimides were formed with moderate to excellent yields. Fortunately, succinimides 3am-3jm were proven to have good functional group tolerance. A range of substituents involving halogen atom, electron-withdraw and electron-donating groups all worked well in yields from 44% to 99%. However, cyclopropane-substituted succin-imide 3an were synthesized in low yield. The structure of 3an was confirmed by single crystal X-ray diffraction (Scheme 5). Apart from this, glutarimides 3ao-3jo and 3ap could be well synthesized. It should be noted that more ring-strain hindered malonimide 3aq could also be prepared.

|
Download:
|
| Scheme 4. Substrate scope of alkyl-cyanoesters 2. Reactions were performed on 0.4 mmol scale; isolated yield. | |
{kind=link}

|
Download:
|
| Scheme 5. The X-ray structure of the N-substituted imide 3an (CCDC: 2191460) | |
{kind=link}
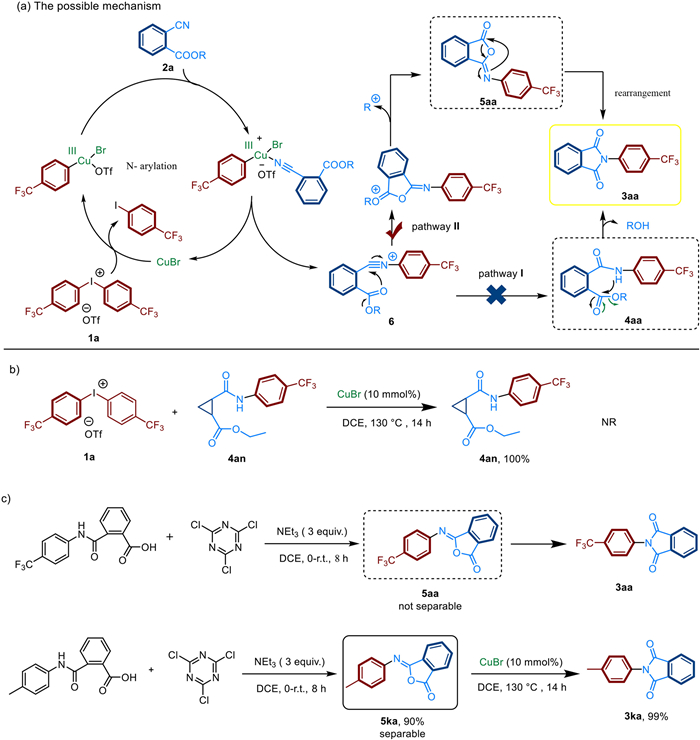
To gain insights into the mechanism of this tandem N-arylation-acylation and rearrangement reaction, a few experi-ments were conducted. Based on the above results and the previous studies in diaryliodonium-involved cascade reactions, we proposed two possible pathways with cyanoesters 2a and diaryliodonium 1a as the model. Firstly, cyanoesters reacted with diaryliodonium salts to form the arylnitrilium cation intermediate 6. One possible pathway Ⅰ (Scheme 6a, pathway Ⅰ): this cation intermediate 6 was easy to transform to amide intermediate 4aa under transition metal catalysis. In the next step, the product was afforded by an intramolecular cyclization via the nucleophilic attack of amide to ester moiety to. To demonstrate whether this process experi-enced an intermediate amide, the reaction of amide 4an with diaryliodonium salt 1a under the optimal conditions did not produce corresponding products 3an (Scheme 6b). This result excluded the possibility of pathway Ⅰ. The other pathway Ⅱ: the cation intermediate 6 via intramolecular cyclization transformed to isophthalimide intermediate 5aa followed by an intramolecular 1,3-(O-N) acyl transfer rearrangenent to give 3aa. To investigate this pathway, intermediate 5aa was tried to be separated, but it was too active to be isolated and rapidly converted into the final product 3aa. On contrast, intermediate 5ka could be isolated (Scheme 6c). The reaction of isophthalimide intermediate 5ka with diaryliodonium salt 1a was carried out and pleasantly, products 3ka could be further afforded in 99% yield (Scheme 6c). Above results demonstrated that isophthalimide 5 might be crucial intermediate in this reaction.

|
Download:
|
| Scheme 6. Mechanistic studies. | |
{kind=link}
Based on the above-mentioned results, we may come to a plausible reaction mechanism for the reaction between diaryl-iodonium salts and cyanoesters (Scheme 6a, pathway Ⅱ). Initially, diaryliodonium salts 1a was affected by copper salts to generate electrophilic species, Ar-CuⅢ [27-39], which was easily coordinated by cyanoester 2a. Then the cation 6 with high electrophilicity was afforded via elimination of CuBr. Next, the isophthalimide cation intermediate was obtained through the intramolecular nucleophilic addition of the ester to the nitrile group. Immediately after the alkyl cation leaved, the isophthalimide intermediate 5aa was formed. Ultimately, by means of intramolecular 1,3-(O-N) acyl transfer rearrangement [40-44] to afford N-aryl-substituted cyclic amide 3aa.
In summary, we have developed a novel and efficient strategy for cyclic imides via the CuBr-catalyzed tandem N-arylation-acylation reaction. This method has excellent functional group compatibility with the simpler and more accessible cyanoesters and diaryliodonium salts as substrate. A wide range of cyclic imides including phthalimides and unsubstituted cyclic imides are synthesized in good yields (up to 99%). This method has also been successfully applied to the generation of fused cyclic imides including malonimides, succinimides and glutarimides.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 21871158 and 22071129).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107913.
| [1] |
Z. Cheng, X. Zhu, E.T. Kang, K.G. Neoh, Macromolecules 39 (2006) 1660-1663. DOI:10.1021/ma0520601 |
| [2] |
J.A. Letizia, M.R. Salata, C.M. Tribout, et al., J. Am. Chem. Soc. 130 (2008) 9679-9694. DOI:10.1021/ja710815a |
| [3] |
U. Sharma, P. Kumar, N. Kumar, B. Singh, Mini-Rev. Med. Chem. 10 (2010) 678-704. DOI:10.2174/138955710791572442 |
| [4] |
Y.J. Wu, G. Liao, B.F. Shi, Green Synth. Catal. 3 (2022) 117-136. DOI:10.1016/j.gresc.2021.12.005 |
| [5] |
P. Tang, H. Wang, W. Zhang, F.E. Chen, Green Synth. Catal. 1 (2020) 26-41. DOI:10.1016/j.gresc.2020.05.006 |
| [6] |
D.H. Tuo, Y.F. Ao, Q.Q. Wang, D.X. Wang, CCS Chem. 4 (2022) 2806-2815. DOI:10.31635/ccschem.021.202101366 |
| [7] |
Y.J. Xu, S.W. Leng, C.M. Xue, et al., Angew. Chem. Int. Ed. 46 (2007) 3896-3899. DOI:10.1002/anie.200604607 |
| [8] |
A.A.M. Abdel-Aziz, K.E.H. El Tahir, Y.A. Asiri, Eur. J. Med. Chem. 46 (2011) 1648-1655. DOI:10.1016/j.ejmech.2011.02.013 |
| [9] |
J. Mulchande, R.C. Guedes, W.Y. Tsang, et al., J. Med. Chem. 51 (2008) 1783-1790. DOI:10.1021/jm701257h |
| [10] |
J. Mulchande, R. Oliveira, M. Carrasco, et al., J. Med. Chem. 53 (2010) 241-253. DOI:10.1021/jm901082k |
| [11] |
S. Shau, T. Juang, W. Ting, et al., Polym. Chem. 2 (2011) 2341-2349. DOI:10.1039/c1py00238d |
| [12] |
M.A. Ali, S.M.A.H. Siddiki, K. Kon, J. Hasegawa, K.I. Shimizu, Chem. Eur. J. 20 (2014) 14256-14260. DOI:10.1002/chem.201404538 |
| [13] |
X.R. Li, J. Zhan, Y.S. Li, Macromolecules 37 (2004) 7584-7594. DOI:10.1021/ma049232z |
| [14] |
M.V. Khedkar, S.R. Khan, D.N. Sawant, D.B. Bagal, B.M. Bhanage, Adv. Synth. Catal. 353 (2011) 3415-3422. DOI:10.1002/adsc.201100460 |
| [15] |
P. Wójcik, A.M. Trzeciak, Appl. Catal. A 560 (2018) 73-83. DOI:10.1016/j.apcata.2018.04.043 |
| [16] |
C. Hong, A. Howard, Org. Lett. 12 (2010) 4126-4129. DOI:10.1021/ol101714p |
| [17] |
D.N. Sawant, Y.S. Wagh, K.D. Bhatte, B.M. Bhanage, Eur. J. Org. Chem. 353 (2011) 6719-6724. |
| [18] |
F.H. Ji, J.X. Li, X.W. Li, et al., J. Org. Chem. 83 (2018) 104-112. DOI:10.1021/acs.joc.7b02433 |
| [19] |
D. Ya, T.K. Hyster, T. Rovis, Chem. Commun. 47 (2011) 12074-12076. DOI:10.1039/c1cc15843k |
| [20] |
S. Inoue, H. Shiota, Y. Fukumoto, N. Chatani, J. Am. Chem. Soc. 131 (2009) 6898-6899. DOI:10.1021/ja900046z |
| [21] |
N. Sharmaa, G. Sekar, Adv. Synth. Catal. 358 (2016) 314-320. DOI:10.1002/adsc.201500642 |
| [22] |
S.P. Chavan, B.M. Bhanage, Eur. J. Org. Chem. 80 (2015) 2405-2410. |
| [23] |
Y.Y. Wang, Y. Zhou, M. Lei, et al., Tetrahedron 75 (2019) 1180-1185. DOI:10.1016/j.tet.2019.01.023 |
| [24] |
M. Wang, J.M. Lu, J.P. Ma, Z. Zhang, F. Wang, Angew. Chem. Int. Ed. 54 (2015) 14061-14065. DOI:10.1002/anie.201508071 |
| [25] |
X. Wang, W.F. Xiong, Y.B. Huang, et al., Org. Lett. 19 (2017) 5818-5821. DOI:10.1021/acs.orglett.7b02771 |
| [26] |
B. Wang, D. He, B. Ren, T. Yao, Chem. Commun. 56 (2020) 900-903. DOI:10.1039/C9CC08438J |
| [27] |
Y. Wang, C. Chen, J. Peng, M. Li, Angew. Chem. Int. Ed. 52 (2013) 5323-5327. DOI:10.1002/anie.201300586 |
| [28] |
X. Su, C. Chen, Y. Wang, et al., Chem. Commun. 49 (2013) 6752-6754. DOI:10.1039/c3cc43216e |
| [29] |
Y. Wang, C. Chen, S. Zhang, et al., Org. Lett. 15 (2013) 4794-4797. DOI:10.1021/ol402164s |
| [30] |
X. Pang, C. Chen, X. Su, M. Li, L. Wen, Org. Lett. 16 (2014) 6228-6231. DOI:10.1021/ol503156g |
| [31] |
J. Peng, C. Chen, J. Chen, et al., Org. Lett. 16 (2014) 3776-3779. DOI:10.1021/ol501655g |
| [32] |
J. Chen, C. Chen, J. Chen, G. Wang, H. Qu, Chem. Commun. 51 (2015) 1356-1359. DOI:10.1039/C4CC08363F |
| [33] |
G. Wang, C. Chen, J. Peng, Chem. Commun. 52 (2016) 10277-10280. DOI:10.1039/C6CC05735G |
| [34] |
J. Sheng, Y. Wang, X. Su, R. He, C. Chen, Angew. Chem. Int. Ed. 56 (2017) 4824-4828. DOI:10.1002/anie.201700696 |
| [35] |
G.X. Liu, C. Wua, B.F. Chen, R. He, C. Chen, Chin. Chem. Lett. 29 (2018) 985-988. DOI:10.1016/j.cclet.2017.11.046 |
| [36] |
T.X. Yan, C. Chen, L.R. Wen, Chin. J. Org. Chem. 41 (2021) 3660-3667. DOI:10.6023/cjoc202104007 |
| [37] |
Y.X. Wu, C. Wu, F. Wang, C. Chen, New J. Chem. 46 (2022) 945-949. DOI:10.1039/D1NJ05433C |
| [38] |
Y.R. Niu, C.Y. Cao, C.X. Ge, H.M. Qu, C. Chen, Chin. Chem. Lett. 33 (2022) 1541-1544. DOI:10.1016/j.cclet.2021.09.004 |
| [39] |
C.Y. Cao, Y.R. Niu, Y.C. Jiang, H.M. Qu, C. Chen, Chin. J. Org. Chem. 42 (2022) 2098-2105. DOI:10.6023/cjoc202203020 |
| [40] |
L. Liu, J. Qiang, S.H. Bai, et al., Adv. Synth. Catal. 359 (2017) 1283-1289. DOI:10.1002/adsc.201601403 |
| [41] |
K. Aradi, Z. Novák, Adv. Synth. Catal. 357 (2015) 371-376. DOI:10.1002/adsc.201400763 |
| [42] |
A. Massa, A. Roscigno, P. DeCapraris, R. Filosa, A. DiMola, Adv. Synth. Catal. 352 (2010) 3348-3354. DOI:10.1002/adsc.201000573 |
| [43] |
V. More, A. DiMola, M. Perillo, et al., Synthesis 18 (2011) 3027-3031. |
| [44] |
P. Antico, V. Capaccio, A. DiMola, A. Massa, L. Palombia, Adv. Synth. Catal. 354 (2012) 1717-1724. DOI:10.1002/adsc.201200065 |