 2023, Vol. 34
2023, Vol. 34 


b Laboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China;
c Innovation Center for Marine Drug Screening & Evaluation Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266003, China;
d Department of Biological Engineering, College of Chemical and Biological Engineering, Shandong University of Science and Technology, Qingdao 266590, China
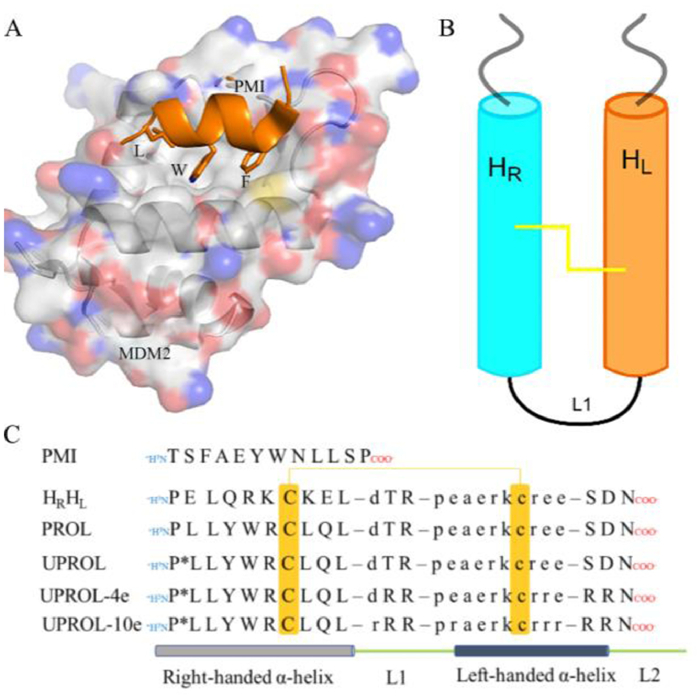
De novo designed proteins could prominently go beyond nature to obtain novel scaffolds by introducing non-canonical backbones and unnatural amino acids [1]. These in silico designed restrained mini-proteins exhibit substantial enzyme and thermal stability, whereas their usage as scaffolds for bioactive mini-protein design are seldom reported. In this study we designed a disulfide-crosslinked, two-helical, hetero-chiral mini-protein for targeting MDM2 from a de novo designed hyperstable constrained peptide (PDB code: 5KX0). The oncoprotein MDM2 negatively regulates the stability and activity of p53, a tumor suppressor protein, and is proved to be an essential molecular target for anti-tumor therapy [2-5]. Over-expression of MDM2 in tumor cells causes p53 inactivation and tumor survival. Therefore, inhibiting p53-MDM2 interaction is an auspicious method for activating p53. Inhibiting p53-MDM2 interaction with different classes of designed compounds could lead to p53-mediated cell-cycle arrest or apoptosis in p53-positive tumor cells in vitro and in vivo [6,7]. Residues from the peptide of p53 transaction domain or p53-MDM2/MDMX inhibitor (PMI) that are critical for MDM2/MDMX binding have been grafted into the naturally occurring restrained peptide for design of competitive inhibitors of the p53-MDM2/MDMX interactions (Fig. 1A) [6,8].

|
Download:
|
| Fig. 1. Structure of MDM2 bound with PMI and the conceived topology of HRHL and its analogues. (A) Crystal structure of MDM2 bound with PMI. (B) The conceived topology of peptide HRHL. (C) Sequences of PMI, HRHL and its analogues. The HRHL topology contains a right-handed helix consisting of L-amino acids (light blue cylinder), a left-handed helix consisting of D-amino acids (orange cylinder), and a disulfide bond (yellow). | |
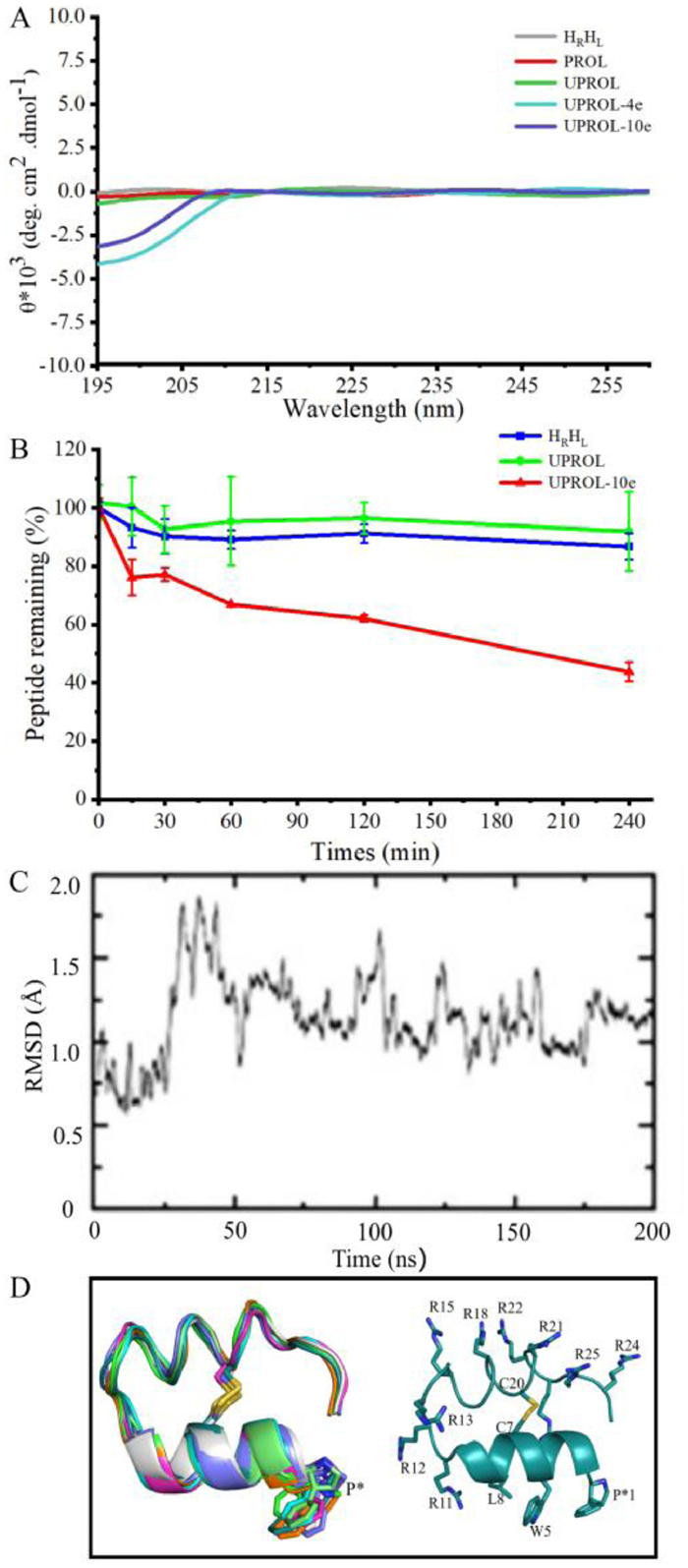
In this study, a 26-residue mini-protein containing two helices was designed to inhibit intracellular p53-MDM2 interactions [9]. To satisfy the bifunctional grafting purposes, a double helices topology was created with one for membrane penetration component design [10] and the other one for MDM2 binding epitope grafting (Figs. 1B and C). Previously de novo designed hyperstable peptide (PDB code: 5KX0) satisfies such a topology except for being a cyclic peptide. Thus, such a peptide was adopted as the initial structure to design the conceived topology by breaking the cyclic bond at Pro 1 and Asn 26 and 200 ns molecular dynamic (MD) simulations were performed to relax the structure. In 200 ns MD simulations the linear peptide HRHL maintained the secondary structure of the 5KX0 except for the C-terminal, which exhibited considerable flexibility and formed random coil in MD. The circular dichroism (CD) spectra for peptide HRHL showed the same characteristic with that of 5KX0, in which merely very weak signal was present between 195 and 260 probably because the L- and D-helical signals largely canceled (Fig. 2A). Besides, the obtained HRHL showed high serum stability despite of the breaking of the cyclic backbone. As shown in Fig. 2B, after four hours there is still 95% of peptide left. The high enzyme stability of the peptide might be originated from the well-formed helix structures and the D-amino acid fragment [1]. Overall, the designed HRHL contains two helical topologies, and is resistant to enzyme digestion. Therefore, it is a suitable scaffold for bifunctional epitope grafting.

|
Download:
|
| Fig. 2. Conformation and stability of the designed peptides. (A) The circular dichroism (CD) spectrum of the designed peptides. The CD spectra was conducted at room temperature with the length of spectra ranged from 195 nm to 260 nm recorded. Only very weak signals were observed because most of the L- and D-helical signals had been canceled [1]. (B) Serum stability of HRHL (green), UPROL (blue), UPROL-10e (red) measured as percentage of peptide remaining in normal human serum. (C) Molecular dynamic simulation of UPROL-10e. (D) Stimulated structure of UPROL-10e. | |
Through bifunctional epitope grafting, critical residues for the binding affinity of PMI were grafted into the N-terminal canonical α-helix consisting of L-amino acids, meanwhile four L- and six D-Arg residues were grafted into the non-canonical α-helix and random coil of the C-terminal, respectively using Rosseta, and then PROL was designed (Fig. 1C) [11]. In consideration of the contribution of the Pro at the N-termini to sustaining the peptide backbone stability and the Phe at the corresponding position of PMI or p53 transaction domain to interacting with the hydro-phobic cleft of MDM2 binding site, an unnatural amino acid UPROL was designed (Fig. 1C). The UPROL is a derivative of PROL with Cβ of the Pro 1 substituted by an aromatic ring. Such a residue can be considered as a hybrid amino acid between Pro and Phe. An initial superimposition of the designed peptide with the PMI-MDM2 crystal structure (PDB code: 3EQS) [12] indicated that side chains for Leu, Trp and Phe at PMI can be well overlapped with the corresponding side chains of Leu, Trp and Pro at the right-handed helix consisting of L-amino acids. For investigation of the influences of the positive net charges to the cellular penetration capacity [10], which in turn can directly affect the cytotoxicity of the designed peptide, we also designed UPROL-4e and UPROL-10e, respectively (Fig. 1C).
Molecular dynamic simulations of the UPROL-10e suggested that the backbone of the peptide maintained stable all through the 200 ns MD even for the N-, C-termini (Figs. 2C and D). The residue at the C-termini forms hydrogen bonds and salt-bridges with the designed UPROL-10e at the N-termini significantly contributing to the stability of the N-, C-termini. In the CD spectra, only minor signal appeared between 195 and 208 nm for UPROL-4e and −10e, while no signal was identified between 208 and 260 nm probably due to the L- and D-helical signals counteracting with each other (Fig. 2B). Thus, CD spectra analysis supports that UPROL, UPROL-4e, and UPROL-10e all maintain the HRHL topology, despite of grafting of varied number of L- or D-amino Arg to the right- and left-handed helices.
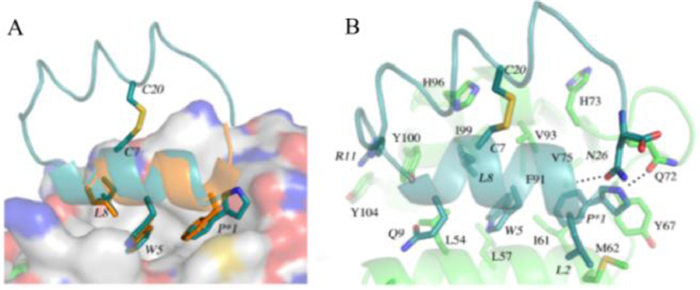
A surface plasmon resonance (SPR) based competition assay was performed to test the binding affinity of the designed peptides (Table 1). PMI has a binding affinity of 17.30 ± 2.58 nmol/L that is consistent with the results given by Pazgier et al. [12]. The binding affinity of UPROL-10e is about 50 times higher than that of PMI, while the binding affinity of UPROL-4e and UPROL is comparable to PMI. By contrast, the PROL had undetectable affinity with MDM2 probably due to the lacking of the aromatic ring at the Cβ of the Pro 1. For understanding the interaction mechanism between the designed peptides with the N-terminal domain of MDM2, computational docking of the highest potent affinity peptide URPOL-10e to the crystal structure of the N-terminal domain of MDM2 was performed using z-dock in Discovery Studio, followed by 50 ns MD simulations in AMBER16 [13]. As shown in Fig. 3A, the right-handed helix of UPROL-10e bound with MDM2 is in similar binding mode to PMI, with side chains of L, W and P*1 oriented to the hydrophobic cleft (Fig. 3A). Interestingly, the aromatic ring at the Cβ of P*1 is well overlapped with the corresponding side chain of the Phe at PMI forming hydrophobic and van der Waals interactions with the hydrophobic cleft of the MDM2 (Fig. 3B). Thus, the absence of the aromatic ring at the PROL is responsible for its undetectable binding affinity to the N-terminal domain of MDM2. Besides, the R11 formed cation-π interaction with Y100 of MDM2, while such an interaction is absent at UPROL and UPROL-4e, which might be responsible for their 10 times less binding affinity to the MDM2 than the UPROL-10e.
|
|
Table 1 The dissociation equilibrium constants (KD) for the designed peptides with the N-terminal domain of MDM2 by SPR assay. |

{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. Docking pose of UPROL-10e with MDM2. (A) Overlapping of the crystal structure of MDM2 bound with PMI (PDB code: 3EQS) with MDM2 bound with UPROL-10e, P* is a derivative of Pro with Cβ substituted by an aromatic ring. (B) UPROL-10e residues of SS-21, Trp5, Leu8 interactions with MDM2. | |
{kind=link}
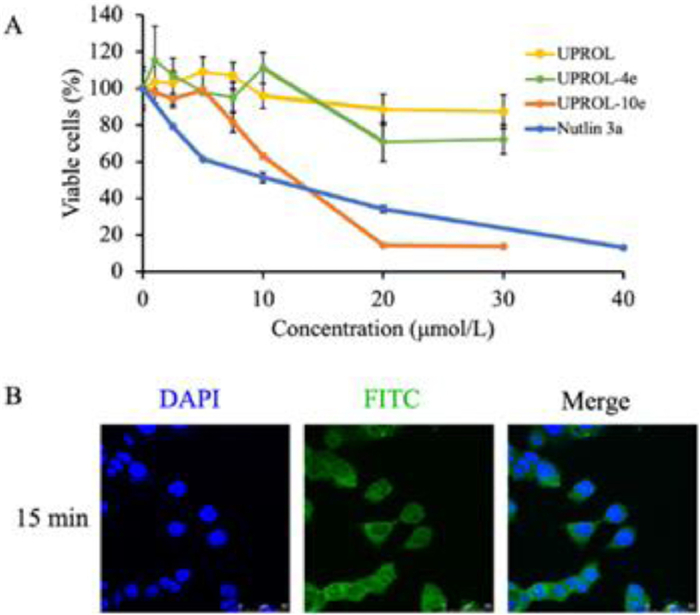
Cellular test results revealed that peptide UPROL and UPROL-4e showed slight cytotoxicity despite of their potent binding affinity on MDM2 (Fig. 4A). Similar results were also observed for the left-handed peptide PMI-D, which was ascribed to their inability to traverse the cell membrane [13]. By contrast, UPROL-10e (IC50 = 12.11 µmol/L) showed comparable activity to the positive control Nutlin-3a (IC50 = 9.37 µmol/L) on p53 expressing HCT116 cells. The presence of cytotoxicity for UPROL-10e is probably originated from its enrichment of positive charges which can enhance its cellular membrane capacity. Using laser confocal detection, we observed the cytoplasmic distribution of FITC labeled UPROL-10e in HCT116 cells after incubation for 15 min (Fig. 4B).

|
Download:
|
| Fig. 4. Inhibitory effect of peptides on the growth of HCT116 human colon cancer cells. (A) HCT116 cells suspended in complete media were seeded in 96-well plates with 5000 cells/well. After 48 h treatment, growth inhibition was measured by MTT assay. (B) Confocal images of the intracellular localization of FITC labeled UPROL-10e at the concentration of 100 µmol/L for 15 min. Scale bar, 50 µm. FITC labeled UPROL-10e was illustrated as green fluorescence and the nuclear was indicated as blue fluorescence, which was stained by DAPI (630×). All the results obtained were in triplicate individual experiments. | |
{kind=link}
In summary, we successfully designed peptide UPROL-10e that possess high binding affinity with the N-terminal domain of MDM2, good cellular penetration capacity as well as potent cytotoxicity against the tumor cells, through bifunctional grating of peptide epitopes to the in silico designed peptide scaffold. The restrained and stable peptide scaffold with novel topology or unnatural amino acid constitution can be designed in silico with atomic-scale precision. These designed peptides scaffolds show great advantage in thermal and enzymatic stability and structural diversity, and functional grafting into these designed stable-structured scaffolds can facilitate the generation of inhibitors for protein-protein interactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 3217110331 and 8212200560) and Major new drug development in Shandong Province (No. 2020CXGC010503).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107871.
| [1] |
G. Bhardwaj, V.K. Mulligan, C.D. Bahl, et al., Nature 538 (2016) 329-335. DOI:10.1038/nature19791 |
| [2] |
P. Chene, Nat. Rev. Cancer 3 (2003) 102-109. DOI:10.1038/nrc991 |
| [3] |
J.C. Marine, M.A. Dyer, A.G. Jochemsen, J. Cell Sci. 120 (2007) 371-378. DOI:10.1242/jcs.03362 |
| [4] |
K.H. Vousden, D.P. Lane, Nat. Rev. Mol. Cell Biol. 8 (2007) 275-283. DOI:10.1038/nrm2147 |
| [5] |
Z. Han, Y. Li, S. Roelle, et al., Bioconjug. Chem. 28 (2017) 1031-1040. DOI:10.1021/acs.bioconjchem.6b00719 |
| [6] |
C. Li, M. Liu, J. Monbo, et al., J. Am. Chem. Soc. 130 (2008) 13546-13548. DOI:10.1021/ja8042036 |
| [7] |
Y. Ji, S. Majumder, M. Millard, et al., J. Am. Chem. Soc. 135 (2013) 11623-11633. DOI:10.1021/ja405108p |
| [8] |
C. Li, M. Pazgier, M. Liu, W.Y. Lu, W. Lu, Angew. Chem. Int. Ed. 48 (2009) 8712-8715. DOI:10.1002/anie.200904550 |
| [9] |
C.E. Laplaza, R.H. Holm, J. Am. Chem. Soc. 123 (2001) 10255-10264. |
| [10] |
S.M. Fuchs, R.T. Raines, Biochemistry 43 (2004) 2438-2444. |
| [11] |
D. Fujiwara, H. Kitada, M. Oguri, et al., Angew. Chem. Int. Ed. 55 (2016) 10612-10615. DOI:10.1002/anie.201603230 |
| [12] |
M. Pazgier, M. Liu, G. Zou, et al., Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 4665-4670. DOI:10.1073/pnas.0900947106 |
| [13] |
M. Liu, M. Pazgier, C. Li, et al., Angew. Chem. Int. Ed. 49 (2010) 3649-3652. DOI:10.1002/anie.201000329 |