 2023, Vol. 34
2023, Vol. 34 


b School of Chemical Sciences, University of the Chinese Academy of Sciences, Beijing 100049, China
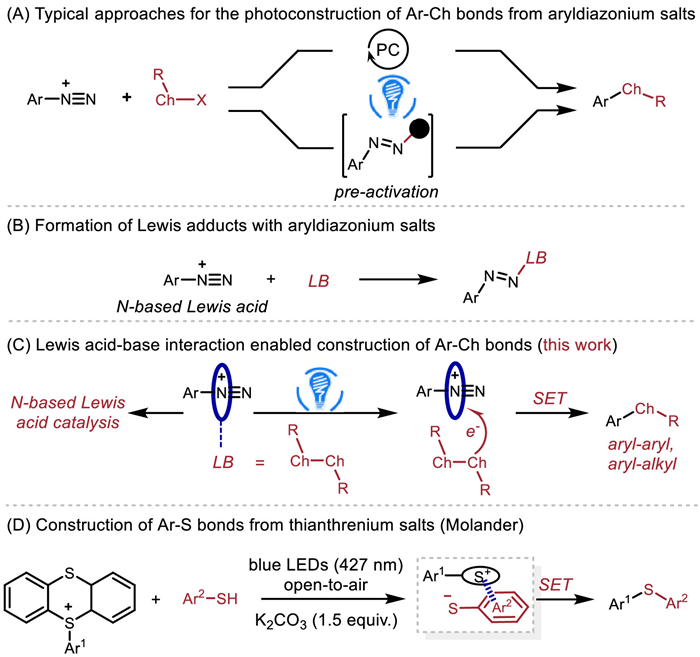
Aryl chalcogen-containing compounds [1-5] which are present in a wide range of pharmaceuticals, agrochemicals and functional materials have always been an inspiration for chemists to design novel strategies for their preparation [6-13]. Traditional methods of forming arylsulfides include the Stadler-Ziegler reaction, which involves the reaction of diazonium salts and thiolates [14-16]. Recently, photoinduced single electron transfer (SET) process has proven useful for the generation of aryl radicals from aryldiazonium salts [17-21]. In the context of the photosynthesis of aryl chalcogenides from aryldiazonium salts and dichalcogenides/thiols, so far photocatalysts, oxidants [22-26] or pre-activation [27,28] of aryldiazonium salts are usually required (Scheme 1A). On the other hand, the Lewis acidity of aryldiazonium salts enables to form Lewis adducts with various nucleophiles and Lewis bases (Scheme 1B) [29-31]. Given the dearth of nitrogen-based Lewis acid catalysts [32-36] and our ongoing research in main-group Lewis acid−base interaction enabled photoreactions [37,38], we set out to explore the possibility of developing Lewis acid catalysis of aryldiazonium salts, and Lewis acidity of aryldiazonium salts enabled the synthesis of aryl chalcogen-containing compounds without using photocatalysts and pre-activation (Scheme 1C). During our preparation of the manuscript, Molander and co-workers reported an interesting electron donor-acceptor (EDA) complex photoactivation strategy for the synthesis of various aryl-aryl sulfides from thianthrenium salts and (hetero)aryl thiols in the presence of potassium carbonate (Scheme 1D) [39].

|
Download:
|
| Scheme 1. Typical approaches for the photoconstruction of C—Ch bonds from aryldiazonium salts or thianthrenium salts vs. our approach. | |
{kind=link}
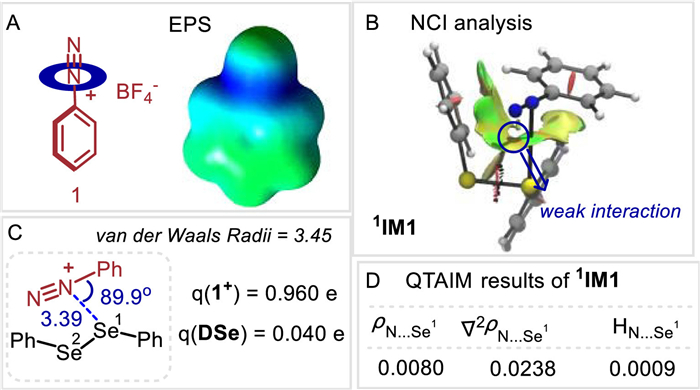
We recently discovered that the diselenides can serve as good chalcogen bonding donors to interact with alkynyl sulfonium salts for the formation of charge-transfer complexes [40]. Guided by this study and our hypothesis, we began our research by investigating the N···Se interaction between aryldiazonium salts and diselenides. Firstly, the electrostatic potential surface (EPS) study of aryldiazonium salt 1 demonstrates that the electron-deficient region is located around of middle nitrogen atom (Fig. 1A). We then carried out non-covalent interaction (NCI) plot index to further analyze the N···Se complex (Fig. 1B) [41,42]. The NCI analysis indicates the attractive interaction in 1 is of noncovalent nature (green surface). Congruent with this view, the binding of diphenyl diselenide 2 to 1 revealed a weak bond (3.39 Å), shorter than the sum of Van der Waals radius (3.45 Å), and a negative charge (0.04 e) is transferred from DSe to 1 component (Fig. 1C). In addition, the quantum theory of atoms (AIM) in molecules provides a way to study chemical bonding [43], in which the chemical bonding generally consists of bond critical points (BCPs) along with the electron density (ρ), Laplacian (∇2ρ), and energy density (H) at BCPs. A small electron density, together with positive Laplacian and energy densities, is widely accepted as a characteristic of donor-accept interaction. For the 1, the electron density (ρ) of 0.0080 a.u. indicates incipient chemical bonding in the range of noncovalent bonding between the practical boundary of a molecule (ρ ≈ 0.001 a.u.) and covalent bonding (ρ > 0.1 a.u.). The positive Laplacian (∇2ρ) of +0.0238 a.u. is also indicative of electron density donation. Moreover, the positive value of H (+0.0009) means the interaction is electrostatic dominant (Fig. 1D). All these results are consistent with the occurrence of weak bonding.

|
Download:
|
| Fig. 1. Computational results. | |
{kind=link}
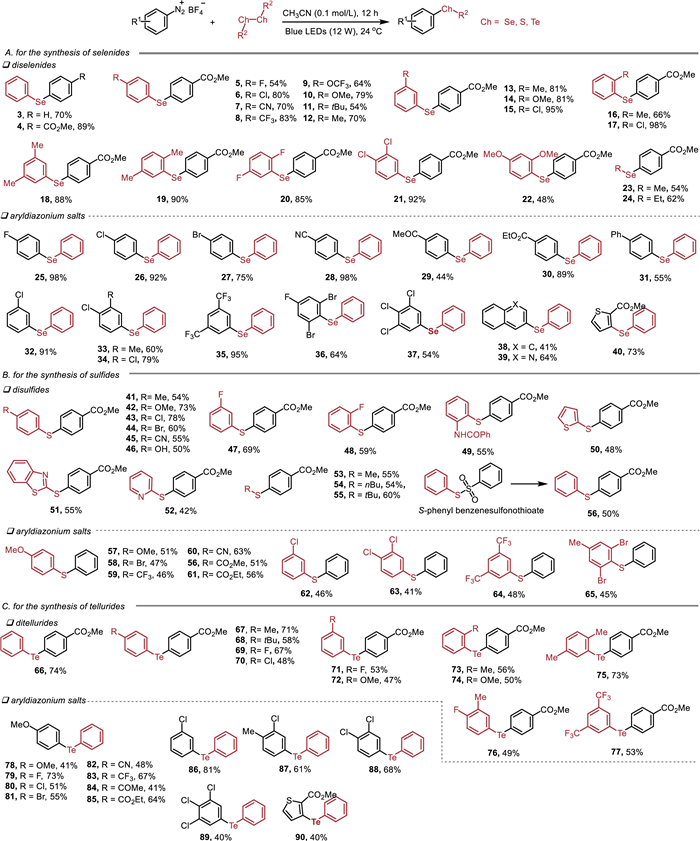
We subsequently assessed the feasibility of the proposed interaction to undergo SET process. To this end, we subjected aryldiazonium salt 1 to diphenyl diselenide 2 under blue light irradiation (12 W). To our delight, the desired product 3 was obtained in 70% yield. In the absence of light, only traces of product 3 were observed. To examine the generality of this reactivity mode. The reaction scope was subsequently conducted and the results are presented in Scheme 2A. Both electron-withdrawing (4-CO2Me, 4-F, 4-Cl, 4-CN and 4-CF3) and electron-donating (4-OCF3, 4-OMe, 4-tBu and 4-Me) groups on the phenyl ring of diselenides were tolerable and provided the desired products 4–12 in moderate to good yields. The meta-(3-Me, 3-OMe and 3-Cl) and ortho-substituents (2-Me and 2-Cl) were also effective (13–17). That was also true for the disubstituted substrates (18–22). Notably, dialkyl diselenides were also transformed to the corresponding products 23 and 24 under these conditions. Then a variety of substituted aryldiazonium salts was evaluated. As expected, substituents changing from single substitution to double or triple substitutions all worked well to give the targeted molecules with satisfactory results (25–37). In addition, naphthalene-, quinoline- and thiophene-substituted diazonium salts also delivered the corresponding products 38–40 in 41%−73% yields.

|
Download:
|
| Scheme 2. Reaction scope. Yields of isolated products are given. | |
{kind=link}
Given the medical applications of sulfides, a convenient protocol for the synthesis of the sulfides was also achieved (Scheme 2B). The electronic properties of the substituents (4-Me, 4-OMe, 4-Cl, 4-Br, 4-CN, 4-OH, 3-F, 2-F and 2-NHCOPh) at disulfides had a limited effect on the yields (41–49). Thiophene-, benzothiazole- and pyridine-substituted disulfides proceeded smoothly to afford the desired products in moderate yields (50–52). The condition was also suitable for dialkyl disulfides (53–55). Notably, S-phenyl benzenesulfonothioate can also be employed in this methodology, affording diphenyl sulfide 56 in 50% yield. Further exploration of the reaction scope showed that various aryldiazonium salts with 4-OMe, 4-Br, 4-CF3, 4-CN, 4-CO2Et, 4-CO2Me and 3-Cl substituents were suitable reaction partners (57–62). Multi-substituted disulfides were also allowed to deliver the target products (63–65).
To demonstrate the synthetic utility of this new strategy, a range of tellurides was then prepared (Scheme 2C). A series of ditellurides bearing electron-donating (Me, tBu and OMe) or electron-withdrawing substituents (F, Cl) at the para-, meta-, or ortho-positions of the benzene ring reacted smoothly and gave the desired products 66–74 in good yields. The reaction of disubstituted ditellurides proceeded well without apparent change in the yields (75–77). The scope of the reaction with respect to the aryldiazonium salts was also examined and different substituted substrates delivered the desired products in moderate to good yields (78–90).
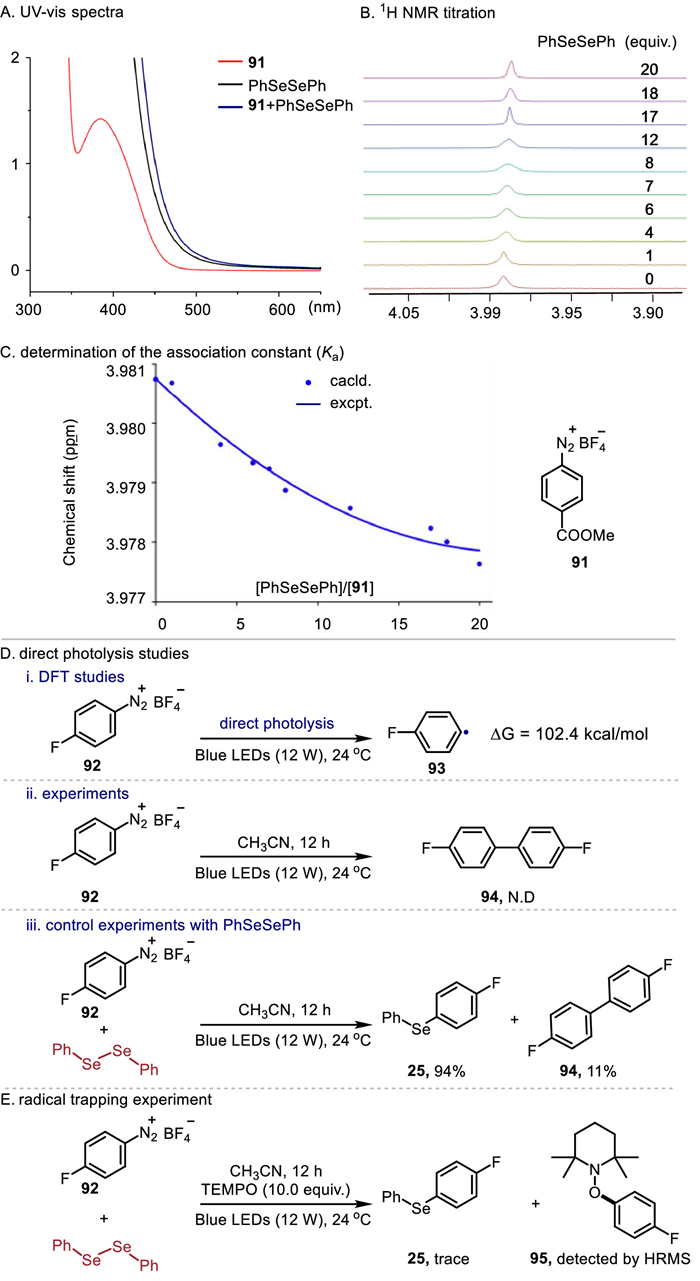
To shed light on the reaction mechanism, we first carried out UV–vis experiments. Upon mixing 91 and 2, a color change from light yellow to orange was observed, and a new absorption band appeared (Fig. 2A), indicating a weak interaction between them. In addition, 1H NMR titration also suggested a weak interaction between 91 and 2, and their association constant (Ka) was calculated as 15.2 L/mol in CD3CN (Figs. 2B and C). Furthermore, we conducted DFT and direct photolysis studies to determine if diazonium salts can photolyze to produce aryl radicals. The DFT studies showed that a minimum of 102.4 kcal/mol of energy is required for 92 to undergo C—N bond homolysis to produce aryl radical. In addition, the calculated maximum absorption wavelength of 92 is 315 nm. In agreement with this view, direct photolysis of aryldiazonium salt 92 does not lead to dimerization product 94, however, dimerization product 94 is obtained in 11% yield in the presence of diselenide (Fig. 2D). Based on these results, it was unlikely that diazonium salts would undergo direct photolysis in this system. Additionally, the radical trapping reaction indicated that an aryl radical had been generated (Fig. 2E). These control experiments revealed that aryl radicals were generated by a weak interaction between diselenides and aryldiazonium salts.

|
Download:
|
| Fig. 2. Mechanistic studies. | |
{kind=link}
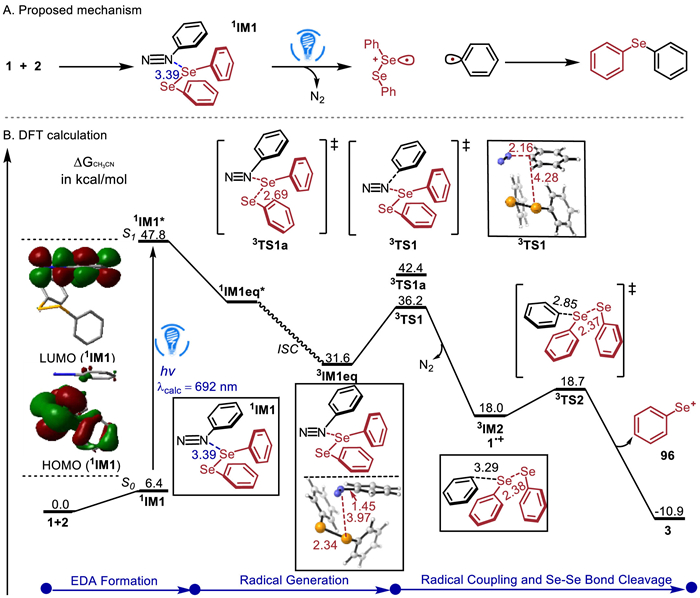
Next, we carried out density functional theory time-dependent DFT (TDDFT) calculations (see Supporting information for computational details) to obtain the reaction pathway using the reaction of 1 with 2 as a representative (Fig. 3). IM1 was predicted to have a maximum absorption wavelength of 695 nm. Thus, irradiation of visible light excites the ground-state IM1 to the first singlet excited state 1IM1*. The examination of HOMO and LUMO of IM1 indicates that the excitation results in the single-electron transfer (SET) from Se to N. Then 1IM1* relaxes to an equilibrium structure 1IM1eq* in the first excited state. Alternatively, 1IM1eq* may convert to the triplet 3IM1eq via an intersystem crossing. Subsequently, the C—N bond cleavage of 3IM1eq via 3TS1 with a low barrier of 4.6 kcal/mol gives phenyl radical 3IM2 and 1•+ (PhSeSePh•+), which is more favorable than Se-Se bond cleavage (3TS1a). The resultant radical 3IM2 attacks 1•+ (2TS2) with a barrier of 0.7 kcal/mol, generating positive ion 96+ and affording product 3. The intermediate 96+ may react with CH3CN to convert into the by-product.

|
Download:
|
| Fig. 3. Computational results. | |
{kind=link}
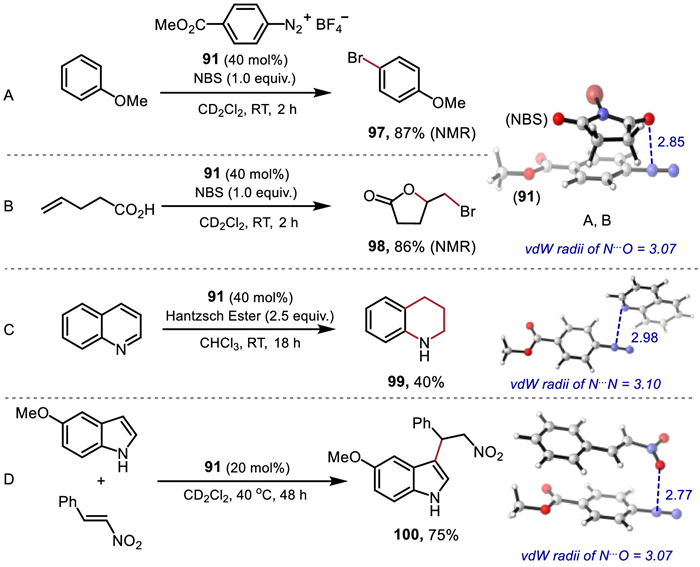
Inspired by the above studies, aryldiazonium salt 91 was investigated as a potential Lewis acid catalyst for the bromination of anisole [44] with NBS by using catalyst loadings of 40 mol% (Fig. 4A). After two hours at room temperature, 87% yield of 97 was observed. Similar reactivity was also observed for the bromolactonization of unsaturated carboxylic acid [44] with NBS, affording the desired product 98 in 86% yield within 2 h (Fig. 4B). In addition, the reaction of quinoline with a Hantzsch ester [45] yielded the transfer hydrogenation product 99 in 40% yield (Fig. 4C). For nitro-Michael addition reaction [46], a catalyst loading of 20 mol% led to the corresponding product 100 in 75% yield (Fig. 4D).

|
Download:
|
| Fig. 4. Reactivity investigations using aryldiazonium salt as Lewis acid catalyst. | |
{kind=link}
In conclusion, various aryl chalcogen-containing compounds can be easily obtained under photoinduced simple and additive-free conditions from aryldiazonium salts and dichalcogenides. In addition, we have extended the nitrogen-containing Lewis acid catalyst club by exploration of the performance of aryldiazonium salts for four benchmark catalytic reactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe acknowledge financial support from the National Natural Science Foundation of China (Nos. 22001248 and 22173103) and the Fundamental Research Funds for the Central Universities and the University of the Chinese Academy of Sciences.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107821.
| [1] |
C.W. Nogueira, G. Zeni, J.B.T. Rocha, Chem. Rev. 104 (2004) 6255-6286. DOI:10.1021/cr0406559 |
| [2] |
M. Feng, B. Tang, S.H. Liang, X. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200-1216. DOI:10.2174/1568026615666150915111741 |
| [3] |
B. Banerjee, M. Koketsu, Coord. Chem. Rev. 339 (2017) 104-127. DOI:10.1016/j.ccr.2017.03.008 |
| [4] |
P. Devendar, G.F. Yang, Top. Curr. Chem. 375 (2017) 82. DOI:10.1007/s41061-017-0169-9 |
| [5] |
W. Hou, H. Xu, J. Med. Chem. 16 (2016) 1200-1216. |
| [6] |
C. Uyeda, Y.C. Tan, G.C. Fu, J.C. Peters, J. Am. Chem. Soc. 135 (2013) 9548-9552. DOI:10.1021/ja404050f |
| [7] |
M.W. Johnson, K.I. Hannoun, Y. Tan, G.C. Fu, J.C. Peters, Chem. Sci. 7 (2016) 4091-4100. DOI:10.1039/C5SC04709A |
| [8] |
M. Jouffroy, C.B. Kelly, G.A. Molander, Org. Lett. 18 (2016) 876-879. DOI:10.1021/acs.orglett.6b00208 |
| [9] |
M.S. Oderinde, M. Frenette, D.W. Robbins, B. Aquila, J.W. Johannes, J. Am. Chem. Soc. 138 (2016) 1760-1763. DOI:10.1021/jacs.5b11244 |
| [10] |
M. Jiang, H. Li, H. Yang, H. Fu, Angew. Chem. Int. Ed. 56 (2017) 874-879. DOI:10.1002/anie.201610414 |
| [11] |
Y. Li, M. Wang, X. Jiang, ACS Catal. 7 (2017) 7587-7592. DOI:10.1021/acscatal.7b02735 |
| [12] |
B. Liu, C.H. Lim, G.M. Miyake, J. Am. Chem. Soc. 139 (2017) 13616-13619. DOI:10.1021/jacs.7b07390 |
| [13] |
L. Pan, M.V. Cooke, A. Spencer, S. Laulhé, Adv. Synth. Catal. 364 (2022) 420-425. DOI:10.1002/adsc.202101052 |
| [14] |
O. Stadler, Ber. Dtsch. Chem. Ges. 17 (1884) 2075-2081. DOI:10.1002/cber.188401702106 |
| [15] |
J.H. Ziegler, Ber. Dtsch. Chem. Ges. 23 (1890) 2469-2472. |
| [16] |
G.E. Hilbert, T.B. Johnson, J. Am. Chem. Soc. 51 (1929) 1526-1536. DOI:10.1021/ja01380a033 |
| [17] |
D.P. Hari, B. König, Angew. Chem. Int. Ed. 52 (2013) 4734-4743. DOI:10.1002/anie.201210276 |
| [18] |
M. Majek, A.J. von Wangelin, Acc. Chem. Res. 49 (2016) 2316-2327. DOI:10.1021/acs.accounts.6b00293 |
| [19] |
C. Raviola, S. Protti, Eur. J. Org. Chem. 2020 (2020) 5292-5304. DOI:10.1002/ejoc.202000143 |
| [20] |
S.S. Babu, P. Muthuraja, P. Yadav, P. Gopinath, Adv. Synth. Catal. 363 (2021) 1782-1809. DOI:10.1002/adsc.202100136 |
| [21] |
F. Mo, D. Qiu, L. Zhang, J. Wang, Chem. Rev. 121 (2021) 5741-5829. DOI:10.1021/acs.chemrev.0c01030 |
| [22] |
M. Majek, A.J. von Wangelin, Adv. Synth. Catal. 49 (2013) 5507-5509. |
| [23] |
X. Wang, G.D. Cuny, T. Noël, Angew. Chem. Int. Ed. 52 (2013) 7860-7864. DOI:10.1002/anie.201303483 |
| [24] |
C. Bottecchia, M. Rubens, S.B. Gunnoo, et al., Angew. Chem. Int. Ed. 56 (2017) 12702-12707. DOI:10.1002/anie.201706700 |
| [25] |
D. Kundu, S. Ahammed, B.C. Ranu, Org. Lett. 16 (2014) 1814-1817. DOI:10.1021/ol500567t |
| [26] |
C. Ghiazza, V. Debrauwer, C. Monnereau, et al., Angew. Chem. Int. Ed. 57 (2018) 11781-11785. DOI:10.1002/anie.201806165 |
| [27] |
L. Blank, M. Fagnoni, S. Protti, M. Rueping, Synthesis 51 (2019) 1243-1252. DOI:10.1055/s-0037-1611648 |
| [28] |
J. Liu, M. Tian, Y. Li, et al., Eur. J. Org. Chem. 2020 (2020) 7358-7367. DOI:10.1002/ejoc.202001386 |
| [29] |
E.R.M. Habraken, N.P. van Leest, P. Hooijschuur, et al., Angew. Chem. Int. Ed. 57 (2018) 11929-11933. DOI:10.1002/anie.201806913 |
| [30] |
E.R.M. Habraken, A.R. Jupp, J.C. Slootweg, Synlett 30 (2019) 875-884. DOI:10.1055/s-0037-1612109 |
| [31] |
F.G. Zhang, Z. Chen, C.W. Cheung, J.A. Ma, Chin. J. Chem. 38 (2020) 1132-1152. DOI:10.1002/cjoc.202000270 |
| [32] |
Y. Tulchinsky, M.A. Iron, M. Botoshansky, M. Gandelman, Nat. Chem. 3 (2011) 525-531. DOI:10.1038/nchem.1068 |
| [33] |
A. Pogoreltsev, Y. Tulchinsky, N. Fridman, M. Gandelman, J. Am. Chem. Soc. 139 (2017) 4062-4067. DOI:10.1021/jacs.6b12360 |
| [34] |
J. Zhou, L.L. Liu, L.L. Cao, D.W. Stephan, Angew. Chem. Int. Ed. 57 (2018) 3322-3326. DOI:10.1002/anie.201713118 |
| [35] |
I. Avigdori, A. Pogoreltsev, A. Kaushanski, N. Fridman, M. Gandelman, Angew. Chem. Int. Ed. 59 (2020) 23476-23479. DOI:10.1002/anie.202008798 |
| [36] |
M. Mehta, J.M. Goicoechea, Angew. Chem. Int. Ed. 59 (2020) 2715-2719. DOI:10.1002/anie.201915547 |
| [37] |
Q. Liu, Y. Lu, H. Sheng, et al., Angew. Chem. Int. Ed. 60 (2021) 25477-25484. DOI:10.1002/anie.202111006 |
| [38] |
H. Sheng, Q. Liu, F. Chen, Z.X. Wang, X.Y. Chen, Chin. Chem. Lett. 33 (2022) 4298-4302. DOI:10.1016/j.cclet.2022.01.028 |
| [39] |
M.J. Cabrera-Afonso, A. Granados, G. Molander, Angew. Chem. Int. Ed. 61 (2022) e202202706. |
| [40] |
Y. Lu, Q. Liu, Z.X. Wang, X.Y. Chen, Angew. Chem. Int. Ed. 61 (2022) e202116071. |
| [41] |
E.R. Johnson, S. Keinan, P. Mori-Sánchez, et al., J. Am. Chem. Soc. 132 (2010) 6498-6506. DOI:10.1021/ja100936w |
| [42] |
J. Contreras-García, E.R. Johnson, S. Keinan, et al., J. Chem. Theory Comput. 7 (2011) 625-632. DOI:10.1021/ct100641a |
| [43] |
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580-592. DOI:10.1002/jcc.22885 |
| [44] |
R. Weiss, E. Aubert, P. Pale, V. Mamane, Angew. Chem. Int. Ed. 60 (2021) 19281-19286. DOI:10.1002/anie.202105482 |
| [45] |
S. Benz, J. López-Andarias, J. Mareda, N. Sakai, S. Matile, Angew. Chem. Int. Ed. 56 (2017) 812-815. DOI:10.1002/anie.201611019 |
| [46] |
P. Wonner, A. Dreger, L. Vogel, E. Engelage, S.M. Huber, Angew. Chem. Int. Ed. 58 (2019) 16923-16927. DOI:10.1002/anie.201910639 |