 2023, Vol. 34
2023, Vol. 34 


b School of Pharmacy, Hubei Engineering Research Center of Traditional Chinese Medicine of South Hubei Province, Hubei University of Science and Technology, Xianning 437100, China
Protein kinases are critical signaling conduits in cells, and their dysregulation is ordinary in human diseases such as cancer, autoimmune diseases, diabetes, cardiovascular disease, and neurological disorders [1]. Since imatinib was approved in 2001, the United States Food and Drug Administration has approved 76 kinase inhibitors [2]; currently, many kinase inhibitors are in the preclinical and clinical development phases. While some small-molecule kinase inhibitors have been successfully applied as drugs, the majority remain undeveloped, requiring a further understanding of their mechanisms and side effects to tap into their potential fully [3]. For example, iCDK9, a well-characterized, highly selective inhibitor of CDK9 kinase, can potently suppress the kinase activity of CDK9 and cause genome-wide RNA Pol Ⅱ pausing, ultimately leading to cancer cell apoptosis. Using an in vitro AlphaScreen-based kinase assay, iCDK9 could potently inhibit the CDK9-CycT1 catalytic activity with an IC50 value below the detection limit of 0.4 nmol/L, exhibiting at least 600-fold lower activity toward CDK1-CycB, CDK2-CycA, CDK4-CycD1, CDK7-CycH-MAT1, and CDK8-CycC [4]. Current small molecule CDK9 inhibitors, like iCDK9, are reversible and require continuous target occupancy to maintain CDK9 inhibition. In previous studies, we found that iCDK9 could inhibit the expression of most genes, but the constant inhibition of CDK9 would increase the expression of MYC and other proto-oncogenes [4]. Therefore, although iCDK9 has highly selective against CDK9, its clinical efficacy has yet to be established. To further develop the potential of iCDK9, it is necessary to understand other potential targets or mechanisms of iCDK9 with new strategies.
Recent advances in chemical proteomics have made it a powerful technology to identify the protein targets of drugs and other bioactive small molecules [5-9]. In general, chemical proteomics depends on the design and use of activity-based molecular probes linked to a reporter tag, which could facilitate the enrichment and identification of binding proteins. The most commonly used tags for target identification are biotin tags, which allow the tag-labeled proteins to be easily enriched with streptavidin beads precisely [10-13]. Unlike the biotinylated probes, the emerging PROTAC bifunctional molecules can induce the degradation of target proteins by exploiting the intracellular ubiquitin-proteasome system. PROTAC is a potent technology for protein degradation, widely used as a biological tool to develop therapeutic molecules with the potential of clinical application value [14-17]. A typical PROTAC molecule is bifunctional by combining a target-selective ligand and a specific E3 ligase recruiting ligand via a linker [18]. The PROTAC bifunctional molecules can thus recruit the E3 ligase onto target proteins and induce ubiquitination and protein degradation via the proteasome [14]. So, the combination of PROTAC and proteomics might supply the unknown target information of small molecular compounds. For instance, Nathanael S. Gray et al. designed a multi-kinase degrader by conjugating a 2,4-diaminopyrimidine scaffold of TAE684 with a carbon-binding ligand and used an unbiased, multiplexed quantitative proteomic approach employing tandem mass tag (TMT) reagents to discover 28 kinases, including nine members of the CDK family, as degradable targets [1]. In 2020, they employed chemo-proteomics to build an experimental map of degradable kinases to accelerate chemical probe development and drug discovery [19]. Hence, the PROTAC technique with quantitative chemical proteomics methods expands the chemical proteomics toolbox and might be an alternative strategy for target identification in drug discovery. On the other hand, stable isotope labeling using amino acids in cell culture (SILAC) is a popular and powerful method for quantitative proteomics, identifying and quantifying relative differential protein changes based on the protein abundance on mass spectrometric analysis [3,20]. Therefore, we developed the idea that the PROTAC-SILAC approach (Fig. 1) could be used to identify previously unknown targets of iCDK9 and reveal the underlying pharmacological mechanisms.

|
Download:
|
| Fig. 1. Scheme of PROTAC coupled with SILAC quantitative chemical proteomic experiments to identify protein targets. PROTAC-SILAC approach: Two populations of cells were cultured in “light” (normal) amino acids medium and “heavy” amino acids, respectively. After being treated with DMSO or control probes in light cells and PROTAC probes in heavy cells, the two populations were mixed at a 1:1 ratio, affinity enriched, digested by trypsin, and analyzed through LC-MS/MS. | |
To develop a potent CDK9 degrader, we first designed iCDK9-based PROTAC molecules according to the molecular docking analysis of iCDK9 with the CDK9 ATP-binding pocket (PDB: 5D1J). The molecular docking study showed that 1,4-trans-cyclohexanediamine of iCDK9 was exposed to solvents (Fig. 2A). Besides, CRBN-mediated degradation had better degradation capabilities against CDKs than VHL-mediated [21-24]. So we decided to synthesize the bifunctional PROTAC molecules by derivatizing the 1,4-trans-cyclohexanediamine nitrogen of iCDK9 with different linkers conjugated to the ligand of cereblon (CRBN) E3 ubiquitin ligase pomalidomide or lenalidomide (Fig. 2B). Totally, we synthesized five iCDK9-based PROTACs (CD-1-5) with alkyl linkers containing 4–9 atoms to investigate the potential chemical space. The synthesis route and characterization of iCDK9-based PROTAC molecules can be found in Supporting information.

|
Download:
|
| Fig. 2. Design of PROTAC molecules based on the CDK9 inhibitor iCDK9. (A) The best pose of iCDK9 in CDK9 crystal structure (PDB ID: 3BLQ). (B) Chemical structures of iCDK9-based PROTAC molecules (CD 1–5). | |
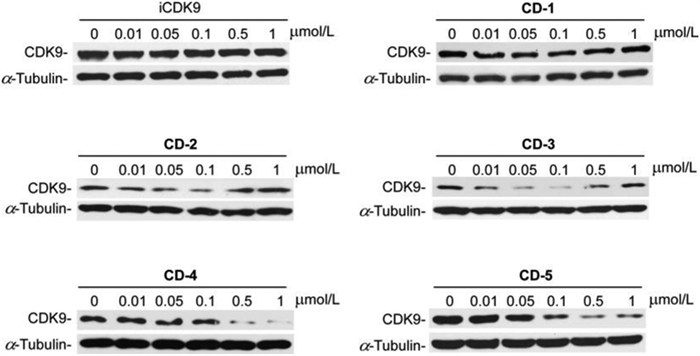
Next, we wanted to screen the effective iCDK9-based PROTAC molecule. We evaluated the ability of compounds CD1-5 to induce the degradation of CDK9 in Jurkat cells (Fig. 3). iCDK9 could not affect the CDK9 protein level, while iCDK9-based PROTAC molecules (except CD-1) showed degradation activity against CDK9. PROTAC molecules with longer linkers such as CD-5 possessed higher degradation capacity, implying that these longer molecules could hold preferable spatial positions for CRBN recruitment toward CDK9. And different from low concentrations, CD-2 and CD-3 failed to induce CDK9 degradation at 1 µmol/L, demonstrating a ‘hook effect’ [25]. Compound CD-4 had the same linker length as CD-3, but its degradation ability was weaker, indicating that pomalidomide recruited E3 ubiquitin ligase better than lenalidomide. Among these CRBN-recruiting PROTACs, CD-5 with the conjugation of iCDK9 and pomalidomide by amide linker demonstrated the best degradation efficacy. CD-5 induced nearly 72% degradation of CDK9 at 0.1 µmol/L and 99% at 0.5 µmol/L in Jurkat cells (Fig. 3 and Fig. S1 in Supporting information).

|
Download:
|
| Fig. 3. PROTAC molecules with different linkers promote CDK9 degradation. The synthesized PROTAC molecules with different linkers were employed for Jurkat cells treatment, and the indicated protein levels were determined by Western blot. | |
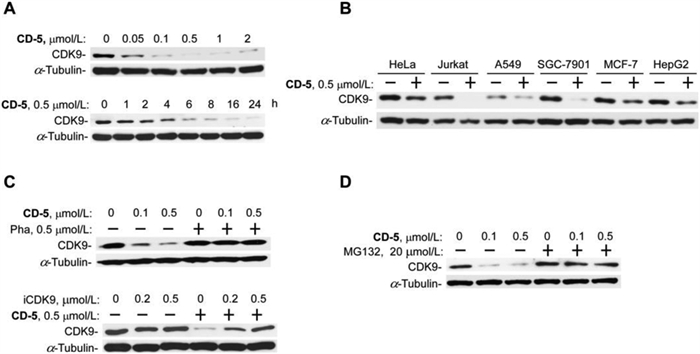
Subsequently, CD-5 performed dose-response and time-course studies on the induction of CDK9 degradation. As shown in Fig. 4A, CD-5 showed dose- and time-dependent CDK9 degradation activity. Time-lapse analysis showed that CD-5 effectively reduced the CDK9 protein level at roughly 2 h and achieved near-complete CDK9-elimination within a 6 h treatment in Jurkat cells. Besides Jurkat cells, CD-5 also significantly induced CDK9 degradation in other cancer cell lines, including HeLa, A549, SGC-7901, MCF-7, and HepG2 (Fig. 4B). These data demonstrated that CD-5 was a very potent and effective CDK9 degrader. Furthermore, we investigated the mechanism of action of CD-5 in inducing CDK9 protein degradation in the Jurkat cell line (Figs. 4C and D). It showed that iCDK9 and pomalidomide could competitively inhibit the degradation effect of CD-5 (Fig. 4C). Similarly, the CDK9 degradation induced by CD-5 could be deterred by the proteasome inhibitor MG-132, consistent with the fact that PROTAC-induced protein degradation was proteasome-dependent (Fig. 4D). The above results implied the ubiquitin-proteasome system mediated the CDK9 degradation effect of CD-5.

|
Download:
|
| Fig. 4. Verification of CD-5 as a CDK9 degrader. (A) CD-5 induces CDK9 degradation in a dose- and time-dependent manner. Jurkat cells were treated with CD-5 at different concentrations for 24 h or treated with 0.5 µmol/L CD-5 at various times as indicated. The whole-cell extracts were prepared and subjected to Western blot. (B) CD-5 promotes CDK9 degradation in various types of cancer cells. Other cancer cells were treated with 0.5 µmol/L CD-5 for 24 h, and whole-cell extras were subjected to Western blot. CD-5 induces CDK9 degradation in a cereblon-based and proteasome-dependent mechanism. Jurkat cells were pretreated with Pha, or iCDK9 (C), or MG132 (D) for 2 h, then incubated with CD-5 for 24 h, and the whole-cell extracts were examined by Western blot. | |
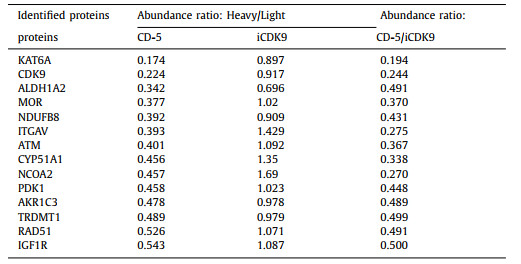
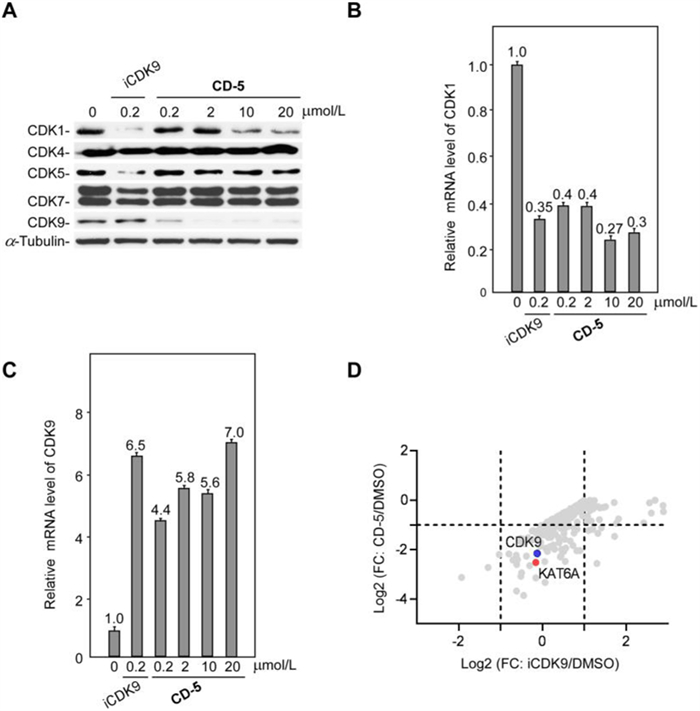
We then examined the responses of other CDKs to CD-5 and found that CD-5 selectively induces the degradation of CDK9 (Fig. 5A). After CD-5 or iCDK9 treatment, the decrease of CDK1 protein could be detected by western blotting. The qRT-PCR results showed that CD-5 could decrease CDK1 mRNA level, indicating that the CD-5-induced reduction in CDK1 protein level resulted from decreased CDK1 mRNA but not protein degradation (Fig. 5B). As a control, the mRNA level of CDK9 did not decrease after CD-5 treatment, which was consistent with our previous result that CD-5 induces CDK9 protein degradation (Fig. 5C). Our data suggested that CD-5 was a potent PROTAC bifunctional molecule of iCDK9. So, to explore the previously unknown targets of iCDK9 and the other underlying mechanisms of action, we selected CD-5 as a chemical probe and used SILAC technology to detect differences in protein abundance among CD-5-treated samples, vehicle samples, and iCDK9-treated samples. In the PROTAC-SILAC experiment, we chose 500 nmol/L as the treatment concentration because the maximal degradation effects were observed at this concentration. Meanwhile, a 12 h-treatment was selected to capture the primary CRBN-dependent degradation response. Upon CD-5 treatment, 14 out of 6998 identified proteins in Jurkat cells were significantly downregulated by more than 50% based on quantification of two or more peptides (false discovery rate [FDR] adjusted P-value < 0.05) (Fig. 5D). Among them, CDK9 was the only CDK to exhibit more than two-log-fold significant downregulation, further confirming the selectivity of CD-5 for CDK9 (Table 1 and Table S1 in Supporting information). Notably, the non-kinase protein KAT6A was most efficiently degraded, with a more than 85% decrease in protein expression (Table 1).

|
Download:
|
| Fig. 5. CD-5 selectively induces degradation of CDK9. (A) Jurkat cells were treated with iCDK9 or CD-5 for 24 h, then the indicated proteins’ levels were determined by Western blot. (B, C) CD-5 decreased the mRNA level of CDK1 but not CDK9. After 24 h treatment with iCDK9 or CD-5, the mRNA levels of CDK1 (B) and CDK9 (C) were measured by qRT-PCR in Jurkat cells. Error bars represent mean ± SD from three separate measurements. (D) Log fold-change in the abundance of proteins as measured using SILAC-based proteomics following a 12 h treatment of Jurkat cells with CD-5 (500 nmol/L) versus iCDK9 (500 nmol/L). Complete data is available in Supporting information. | |
|
|
Table 1 CD-5 significantly degrades proteins in Jurkat cells based on SILAC proteomics. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
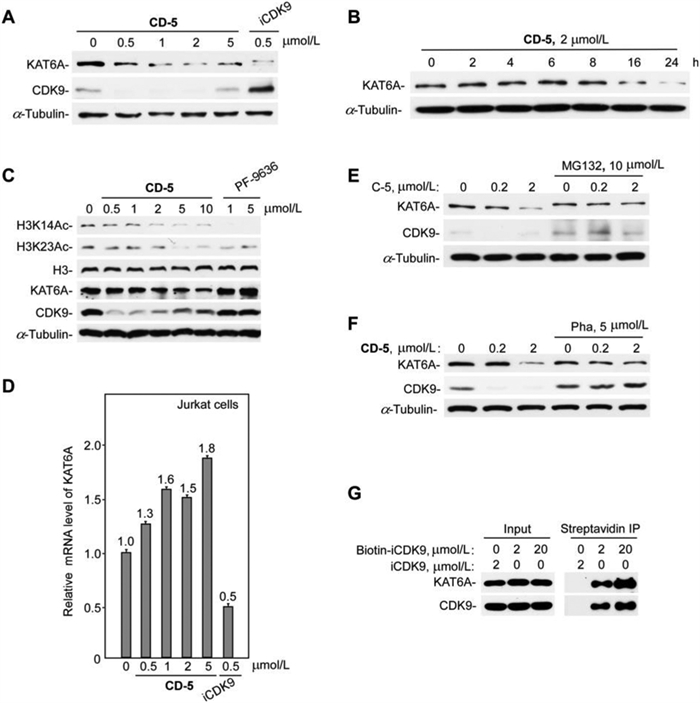
Lysine acetyltransferases (KATs) can acetylate histones, which is essential for chromatin organization and function [26,27]. Oncogenes KAT6A (also known as MOZ) and KAT6B (also known as MORF or QKF) are two genes coding for the MYST family of KATs (KAT5–KAT8) [28]. KAT6A plays a vital role in normal hematopoietic stem cells and is one target of recurrent chromosomal translocations that cause acute myeloid leukemia [26,29]. However, no literature reported that the active compound iCDK9 could bind to KAT6A and block its protein functions. So, we further used western blotting to analyze KAT6A protein levels in CD-5-treated Jurkat cells to confirm the degradation of KAT6A detected by mass spectrometry. We found that CD-5 induced the degradation of KAT6A in a dose- and time-dependent manner (Figs. 6A and B). Simultaneously, the CD-5-induced KAT6A degradation dose-dependently decreased the acetylation of H3K14 and H3K23 (Fig. 6C). As iCDK9 alone could also reduce KAT6A protein level (Fig. 6A), the quantitative real-time PCR was performed to examine whether CD-5 or iCDK9 downregulated KAT6A at the transcriptional level. We found that iCDK9 treatment significantly decreased CDK9 mRNA expression while CD-5 treatment slightly influenced the KAT6A mRNA level (Fig. 6D). However, the proteasome inhibitor MG132 could dramatically inhibit the degradation of KAT6A (Fig. 6E), implying that blocking the ubiquitin-proteasome system could hinder the KAT6A degradation induced by CD-5. These data suggested that KAT6A might be a potentially binding target of iCDK9 and its PROTAC bifunctional molecule CD-5.

|
Download:
|
| Fig. 6. KAT6A is a non-kinase target of iCDK9. (A, B) CD-5 induced KAT6A degradation in a dose- and time-dependent manner. After being treated with the compound (CD-5 or iCDK9) at different concentrations for 24 h (A) or 2 µmol/L CD-5 at different times (B), the whole-cell extracts of Jurkat cells were examined by Western blotting. (C) CD-5 decreased the acetylation of H3K14 and H3K23. Jurkat cells were treated with CD-5 or KAT6A inhibitor PF-9636 for 24 h; then, the indicated proteins were measured by Western blot. (D) The mRNA level of KAT6A was determined by qRT-PCR of Jurkat cells after being treated with CD-5 or iCDK9. Error bars represent mean ± SD from three separate measurements. (E, F) The CD-5-induced degradation of KAT6A depends on the ternary complex formation and ubiquitin-proteasome system. Jurkat cells were pretreated with MG132 (E) or pomalidomide (Poma) (F) for 2 h, then incubated with CD-5 for 24 h as indicated. The protein levels were examined by Western blot. (G) The traditional affinity-based approaches by biotinylation of iCDK9 to purify the potential KAT6A target. Whole-cell extracts of Jurkat cells were incubated with Biotin-CDK9 or iCDK9 for streptavidin Immunoprecipitation (IP). The IP products were then subjected to Western blotting analysis. | |
{kind=link}
To further determine whether CD-5 caused KAT6A degradation by forming the ternary KAT6A:PROTAC:CRBN complex, we used pomalidomide to compete with CD-5 to dissociate the ternary complex. Similarly, pomalidomide treatment could reduce the KAT6A degradation mediated by CD-5 (Fig. 6F). These data demonstrated that the formation of the ternary KAT6A:CD-5:CRBN complex played a crucial role in the CD-5-mediated degradation of KAT6A, and KAT6A might be a potential target of compound iCDK9. So, to confirm whether KAT6A was a direct target of iCDK9 in cells, we attempted to synthesize a biotin-modified iCDK9 probe used to pull down KAT6A. As shown in Fig. 6G, both KAT6A and CDK9 could be engaged by biotin-iCDK9 in Jurkat cells. Besides, in the CDK9 knockdown cells, the protein and mRNA level of KAT6A changed slightly, which indicated that KAT6A was not the target gene of CDK9 (Fig. S2 in Supporting information). Thus, we concluded that KAT6A might be a novel non-kinase target of iCDK9.
In this study, we proposed a novel strategy by combining PROTAC technology with SILAC quantitative proteomics to identify potential unknown targets of iCDK9. We designed and synthesized five iCDK9-based PROTACs bifunctional molecules. The PROTACs bifunctional molecule CD-5 showed excellent and selective CDK9-degradation activity with low cell toxicity (Table S2 in Supporting information). The SILAC profiling data showed CD-5 inducing the degradation of KAT6A, and subsequent validation experiments confirmed that KAT6A was a non-kinase target of iCDK9. Together, this work suggests that compound CD-5 represents a suitable iCDK9-based PROTACs bifunctional molecule for revealing CDK9’s biological function and the underlying pharmacological mechanisms of iCDK9. However, the detailed biological targets of iCDK9 should be defined in the future.
Declaration of competing interestAuthors have no conflict of interest to declare.
AcknowledgmentsThis work was supported by grants from the National Key R&D Program of China (Nos. 2018YFA0107303 and 2020YFA0908100), the National Natural Science Foundation of China (Nos. 81773600 and 82102746), the China Postdoctoral Science Foundation (No. 2021M690095), and the Fundamental Research Funds for the Central Universities (No. 20720180051).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107741.
| [1] |
H.T. Huang, D. Dobrovolsky, J. Paulk, et al., Cell Chem. Biol. 25 (2018) 88-99. DOI:10.1007/s40572-018-0180-5 |
| [2] |
P. Cohen, D. Cross, P.A. Janne, Nat. Rev. Drug Discov. 20 (2021) 551-569. DOI:10.1038/s41573-021-00195-4 |
| [3] |
X. Chen, Y.K. Wong, J. Wang, et al., Proteomics 17 (2017) 1600212. DOI:10.1002/pmic.201600212 |
| [4] |
H. Lu, Y. Xue, G.K. Yu, et al., eLife 4 (2015) 06535. |
| [5] |
S. Ziegler, V. Pries, C. Hedberg, H. Waldmann, Angew. Chem. Int. Ed. 52 (2013) 2744-2792. DOI:10.1002/anie.201208749 |
| [6] |
M.H. Wright, S.A. Sieber, Nat. Prod. Rep. 33 (2016) 681-708. DOI:10.1039/C6NP00001K |
| [7] |
S. Klaeger, S. Heinzlmeir, M. Wilhelm, et al., Science 358 (2017) 4368. DOI:10.1126/science.aan4368 |
| [8] |
H.J. Benns, C.J. Wincott, E.W. Tate, M.A. Child, Curr. Opin. Chem. Biol. 60 (2021) 20-29. DOI:10.1016/j.cbpa.2020.06.011 |
| [9] |
G. Li, X. Peng, Y. Guo, et al., Front. Chem. 9 (2021) 761609. DOI:10.3389/fchem.2021.761609 |
| [10] |
X. Zhao, G. Li, S. Liang, J. Anal. Methods Chem. 2013 (2013) 581093. |
| [11] |
M. Pan, Y. Liu, X. Zheng, et al., Chin. Chem. Lett. 32 (2021) 3479-3482. DOI:10.1016/j.cclet.2021.05.057 |
| [12] |
G. Xie, L. Zhu, Y. Song, et al., Chin. Chem. Lett. 32 (2021) 2164-2168. DOI:10.1016/j.cclet.2020.11.064 |
| [13] |
W. Lei, F. Shen, N. Chang, et al., Chin. Chem. Lett. 32 (2021) 190-193. DOI:10.1016/j.cclet.2020.10.049 |
| [14] |
G.M. Burslem, C.M. Crews, Cell 181 (2020) 102-114. DOI:10.1016/j.cell.2019.11.031 |
| [15] |
D.A. Nalawansha, C.M. Crews, Cell Chem. Biol. 27 (2020) 998-1014. DOI:10.1016/j.chembiol.2020.07.020 |
| [16] |
T.K. Neklesa, J.D. Winkler, C.M. Crews, Pharmacol. Ther. 174 (2017) 138-144. DOI:10.1016/j.pharmthera.2017.02.027 |
| [17] |
S.L. Paiva, C.M. Crews, Curr. Opin. Chem. Biol. 50 (2019) 111-119. DOI:10.1016/j.cbpa.2019.02.022 |
| [18] |
D.P. Bondeson, B.E. Smith, G.M. Burslem, et al., Cell Chem. Biol. 25 (2018) 78-87. DOI:10.1016/j.chembiol.2017.09.010 |
| [19] |
K.A. Donovan, F.M. Ferguson, J.W. Bushman, et al., Cell 183 (2020) 1714-1731. DOI:10.1016/j.cell.2020.10.038 |
| [20] |
S.E. Ong, B. Blagoev, I. Kratchmarova, et al., Mol. Cell Proteomics 1 (2002) 376-386. DOI:10.1074/mcp.M200025-MCP200 |
| [21] |
B. Yu, Z. Du, Y. Zhang, et al., Future Med. Chem. 14 (2022) 167-185. DOI:10.4155/fmc-2021-0154 |
| [22] |
F. Zhou, L. Chen, C. Cao, et al., Eur. J. Med. Chem. 187 (2020) 111952. DOI:10.1016/j.ejmech.2019.111952 |
| [23] |
S. Su, Z. Yang, H. Gao, et al., J. Med. Chem. 62 (2019) 7575-7582. DOI:10.1021/acs.jmedchem.9b00871 |
| [24] |
X. Qiu, Y. Li, B. Yu, et al., Eur. J. Med. Chem. 211 (2021) 113091. DOI:10.1016/j.ejmech.2020.113091 |
| [25] |
V.G. Klein, A.G. Bond, C. Craigon, et al., J. Med. Chem. 64 (2021) 18082-18101. DOI:10.1021/acs.jmedchem.1c01496 |
| [26] |
N. Wiesel-Motiuk, Y.G. Assaraf, Drug Resist. Update. 53 (2020) 100729. DOI:10.1016/j.drup.2020.100729 |
| [27] |
A.K. Voss, T. Thomas, BioEssays 40 (2018) 1800078. DOI:10.1002/bies.201800078 |
| [28] |
A.K. Voss, T. Thomas, BioEssays 31 (2009) 1050-1061. DOI:10.1002/bies.200900051 |
| [29] |
B.N. Sheikh, Y. Yang, J. Schreuder, et al., Blood 128 (2016) 2307-2318. DOI:10.1182/blood-2015-10-676072 |