 2023, Vol. 34
2023, Vol. 34 


b Pingshan Translational Medicine Center, Shenzhen Bay Laboratory, Shenzhen 518055, China;
c School of Civil and Environmental Engineering, Harbin Institute of Technology, Shenzhen 518055, China
UBE2C is a member of the E2 ubiquitin-conjugating enzyme family, which plays a key role in cell cycle progression [1,2]. As an essential factor of the cell cycle-regulated ubiquitin ligase anaphase promoting complex/cyclosome (APC/C), UBE2C promotes the degradation of several target proteins along cell cycle progression, particularly mitotic cyclins, during metaphase/anaphase transition [3-6]. It is reported that UBE2C is highly expressed in numerous cancerous cell lines and various primary tumors in comparison with surrounding normal tissues, highlighting its contribution in the mechanisms of carcinogenesis [2,7]. Meanwhile, many of the consequences of UBE2C overexpression, such as increased proliferation, metastasizing, cancer progression and resistance to anticancer drugs are reversed through gene silencing techniques [8,9]. Therefore, UBE2C is considered to be a potential therapeutic target for the treatment of cancer. However, like other E2s, the lack of deep active site clefts and the need to target the protein surface have led to UBE2C being regarded as an “undruggable” target to small molecules [1,10].
Proteolysis targeting chimeras (PROTACs) are a new class of technology that employ targeted protein degradation as a mode of action [11]. Due to the selective degradation mechanism, PROTACs could target not only some certain domain of target protein, e.g. active sites of enzymes in order to compete with their substrates or protein interaction sites to block the binding, but also any site on the target protein surface as long as it provides specificity for the given molecule [12]. This advantage allows PROTACs to target proteins which was viewed as undruggable target [11-13]. The traditional PROTACs are designed based on small molecules, including one small-molecule ligand to bind with the target protein, another small-molecule ligand to recruit an E3 ubiquitin ligase, and an optimal linker to concatenate the two ligands [13]. However, the design and optimization of a high-affinity small molecular ligand for traditional PROTACs are often laborious and time consuming [14]. Furthermore, there are many potential targets those do not have the small molecule binding sites, e.g. UBE2C, needed for traditional PROTACs development [12].
To overcome these problems, the alternative PROTAC approaches has been developed. BioPROTACs (biological PROTACs) derived from PROTACs are a kind of chimeric fusion proteins consisting of a target binder, usually a peptide or a miniprotein, and an E3 ligase function domain [12]. Unlike traditional PROTACs those using small molecules to bridge the target proteins and the E3 ligase, bioPROTACs directly recruit the target proteins by the binder to the fused E3 ligase [15]. It is reported that the bioPROTACs have been successfully used to degrade the classically “undruggable” targets such as β-catenin [16,17], KRAS [18-20], and c-Myc [21], and other target proteins [22-27].
For the rational design of bioPROTACs, the choice of E3 module is essential because it determines the degradation efficiency [15]. However, the current selection of E3 ligases lacks the diversity and the limited pool of routinely used E3 ligases restrict the development of bioPROTACs. So far, only a few E3 ligases have been applicated in bioPROTAC design, mostly derived from mammalian [14]. In fact, bacterial-derived E3 ligases can also be used in target proteins degradation. IpaH9.8 is an E3 ubiquitin ligase enzyme derived from Shigella flexneri, containing an N-terminal leucine-rich repeats (LRR) domain involved in substrate recognition, and a C-terminal conserved novel E3 ligase (NEL) domain [28]. In a proof-of-concept study, the bioPROTACs based on the NEL domain of IpaH9.8 could significantly reduce the protein levels of the GFP and its spectral derivatives as well as different GFP-tagged mammalian proteins those varied in size and subcellular localization [14]. In this study, we explore the use of the bioPROTAC technology based on the NEL domain of IpaH9.8 for targeted ubiquitination and degradation of UBE2C, and demonstrate its efficacy by in vitro and in vivo assays.
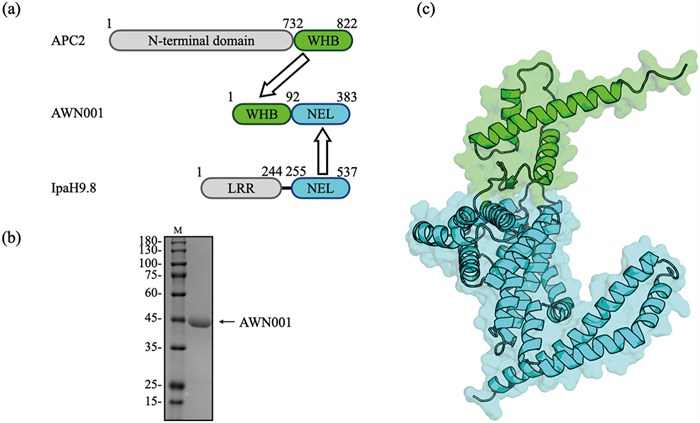
As a starting point, we need to reengineer the IpaH9.8 by replacing its natural substrate recognition LRR domain with a UBE2C binding motif. In general, high-affinity peptides or screened/designed miniproteins, such as antibody mimetics, are recommended as target binders in bioPROTAC design [12]. However, no such binding molecule was reported for UBE2C. Instead, it is reported that a 91 amino acids (aa) motif, so called “winged-helix B” (WHB) domain, of APC2 is responsible for mediating cell cycle ubiquitylation by directly binding with UBE2C [29,30], which makes the APC2’s WHB domain an excellent candidate for the UBE2C recognition module. We fused the WHB domain to the N-terminus of the NEL domain of IpaH9.8 resulting in the bioPROTAC molecule AWN001 (Fig. 1a). Then, the AWN001 was expressed in Escherichia coli, and subsequently purified (Fig. 1b and Figs. S1a-c in Supporting information). The gel filtration chromatography result shows that AWN001 can exist as both monomer and dimer in solution (Figs. S1b and c in Supporting information), which is consistent with our previous research that IpaH9.8 can form a NEL-mediated dimer [28]. Subsequently, the three-dimensional structure of AWN001 was predicted using AlphaFold2 program (Fig. 1c) [31], showing that the structures of WHB domain and NEL domain in AWN001 are almost identical to those in native state (Fig. S1d in Supporting information).

|
Download:
|
| Fig. 1. (a) Schematic representations of the design of the bioPROTAC AWN001 construct. The NEL domains are colored green, the WHB domains are colored cyan, and the LRR domain of IpaH.9.8 and the N-terminal domain of APC2 are colored gray. (b) The Coomassie Brilliant Blue (CBB) stained SDS-PAGE gel of AWN001. First lane, marker (kDa); second lane, purified AWN001. (c) The cartoon model of predicted AWN001 structure with domains colored as described in (a). | |
{kind=link}
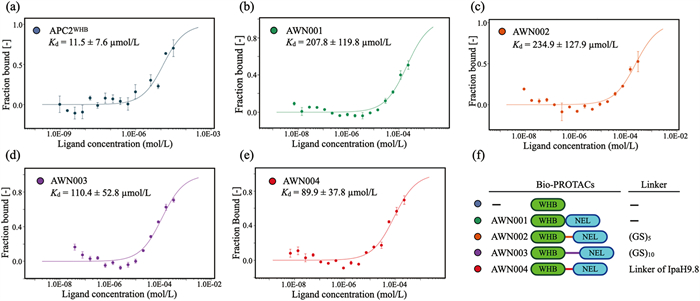
To test whether binding of the WHB domain with the target, UBE2C, was hindered by fusion to the E3 ligase module NEL domain, we tested both isolated WHB domain and bioPROTAC molecule AWN001 for interaction with fluorescent-labeled UBE2C by microscale thermophoresis (MST) (Figs. 2a and b). The result shows that the binding affinity of AWN001 to UBE2C (Kd ~207.8 µmol/L; Fig. 2b) is about 18 times lower than that of isolated WHB domain (Kd ~11.5 µmol/L; Fig. 2a), suggested that fusing to the NEL domain resulted in a decrease in binding affinity of the WHB domain for UBE2C. To improve the target-binding affinity of our bioPROTAC molecule, we turned our attention to the linker connecting the binder module and the E3 ligase module. Indeed, we recently demonstrated that the linker between the substrate recognition domain and E3 ligase domain in IpaH9.8 is critical for its ubiquitination activity for substrate [28]. Moreover, the optimization of linker is often an important step for traditional PROTAC design even through it has never been systematically explored for bioPROTAC action [12]. Three different type linkers, a 10 aa length glycine‑serine repeat linker (GS)5, a 20 aa length glycine‑serine repeat linker (GS)10, and an 11 aa length IpaH9.8-derived linker (GQQNTLHRPLA), were separately introduced to produce bioPROTAC molecules AWN002, AWN003, and AWN004, respectively (Figs. 2c-e). Interestingly, the subsequent MST assays reveal that AWN004 shows the higher affinity to UBE2C (Kd ~89.9 µmol/L; Fig. 2e) than that of AWN002 (Kd ~234.9 µmol/L; Fig. 2c) and AWN003 (Kd ~110.4 µmol/L; Fig. 2d). This result suggested the effects of linker on bioPROTAC target-binding activity are not only dependent on linker length but related by linker sequence (Fig. 2f, Fig. S2 in Supporting information).

|
Download:
|
| Fig. 2. (a-e) Binding analysis of UBE2C to isolated WHB domain or indicated bioPROTACs using MST. The calculated Kd values (n = 3) are labeled, respectively. Errors in Kd represent fitting errors. (f) Schematic representations of the constructs of isolated WHB domain and indicated bioPROTACs with different linkers. The linkers are listed at right. The NEL domains are colored green, and the WHB domains are colored cyan. | |
{kind=link}
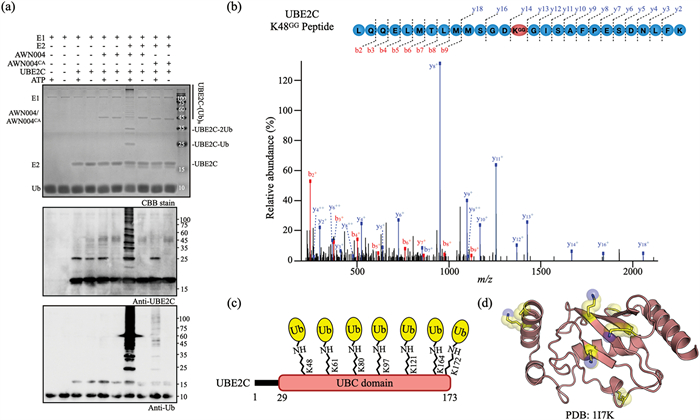
Next, we performed in vitro ubiquitination assays with purified components, including AWN004 as the E3 enzyme and UBE2C as the target (Fig. 3a). UbcH5b was used as the E2 enzyme because it has previously been proved to specifically function with NEL E3 ligase [32]. High molecular weight bands corresponding to ubiquitinated UBE2C were only observed when all pathway components were included in the reaction (Fig. 3a), indicating that the bioPROTAC activity was dependent on a complete ubiquitination pathway. In addition, AWN004 could only recognize UBE2C but not UBE2S, which is also an APC/C related E2 enzyme, as substrate (Fig. S3 in Supporting information), demonstrated the specificity of AWN004 for UBE2C. These results confirm that the NEL domain retained E3 ligase activity in the context of AWN004. Moreover, the AWN004CA, which carries an active cystine to alanine substitution in NEL domain (Fig. S2), failed to conjugate ubiquitin to UBE2C (Fig. 3a), indicating the complete NEL domain is necessary for the substrate ubiquitin modification.

|
Download:
|
| Fig. 3. (a) In vitro ubiquitination assay of UBE2C by AWN004 or AWN004CA. Reaction products were detected by CBB staining (top), anti-UBE2C antibody (middle), and anti-ubiquitin antibody (bottom). (b) A representative MS/MS spectrum of a UBE2C peptide that includes the identified ubiquitination site Lys-48 is depicted. The ubiquitinated lysine residue is labeled with GG. Fractionation of the peptides into b and y ions was performed, and the corresponding peaks are labeled on the spectra. The y axis, relative abundance, was normalized to the most abundant identified peptide fragment. (c) Schematic representation of the UBE2C construct with ubiquitinated lysine residues indicated. The UBC domain of UBE2C is colored red, and the ubiquitins are colored yellow. (d) Mapping of ubiquitinated lysines (yellow; the side chain nitrogen atom colored blue) onto the three dimention structure of UBE2C (red; PDB: 1I7K). | |
{kind=link}
We further investigated the precise modification site of UBE2C after the in vitro ubiquitination assays. Bands on the SDS-PAGE gel corresponding to modified UBE2C by AWN004 (Fig. 3a, top) were excised, digested with trypsin, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Trypsin digestion of a ubiquitinated protein leaves the C terminus of ubiquitin, Gly-Gly, attached to the ubiquitinated lysine residue [33]. We analyzed the MS data and found that ubiquitin was linked to seven lysines of UBE2C: Lys-48, Lys-61, Lys-80, Lys-97, Lys-121, Lys-164, and Lys-172 (Figs. 3b and c, and Figs. S4-S9 in Supporting information). Besides Lys-48, other six lysines are consistent with the location of ubiquitin attachment sites on native UBE2C [34-37]. Further structural analysis revealed that all modified lysines are located in the surface of UBE2C (Fig. 3d).
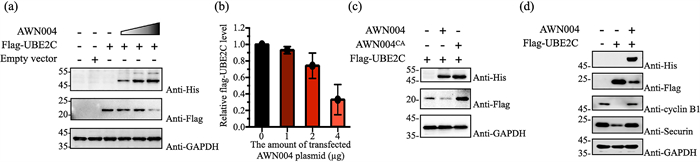
Having successfully demonstrated the in vitro ubiquitination ability of AWN004, we were prompted to investigate whether it could be used to degrade the UBE2C through in mammalian cells. HeLa cells were transiently co-transfected with pcDNA3.1-based plasmids encoding His-tagged AWN004 and FLAG-tagged UBE2C. Forty-eight-hours post-transfection, cellular His-AWN004 and FLAG-UBE2C levels were measured by immunoblotting. When HeLa cells were co-transfected with pcDNA-FLAG-UBE2C and increasing amounts of pcDNA-His-AWN004, the FLAG-UBE2C levels were systematically reduced to as low as 30% of the steady-state levels measured in cells transfected with only the pcDNA-FLAG-UBE2C plasmid, while the levels of a house-keeping protein, GAPDH, were not affected by co-transfections (Figs. 4a and b, Figs. S10a and b in Supporting information). The plasmid encoding AWN004 dosage was correlated linearly with the extent of UBE2C degradation. In contrast, no reduction in UBE2C expression following co-transfection with the plasmids encoding mutant AWN004CA (Fig. 4c). Moreover, co-transfected with pcDNA-FLAG-UBE2C and pcDNA-His-AWN004 could block the turnover of UBE2C native substrates including cyclin B1 and securin in HeLa cells which was transfected with only the pcDNA-FLAG-UBE2C plasmid, strongly validated the degradation effect of AWN004 for the exogenous UBE2C (Fig. 4d). In addition, treating the cells with MG132, a proteasome inhibitor, or TAK-243, a UAE inhibitor, rescued the UBE2C degradation (Fig. S10b), supporting that UBE2C was targeted by AWN004 for degradation in a proteasome-dependent manner.

|
Download:
|
| Fig. 4. (a) Immunoblots of extracts from HeLa cells transfected with pcDNA-FLAG-UBE2C (2 µg) alone or co-transfected with pcDNA-UBE2C (2 µg) and pcDNA-His-AWN004. The triangle indicates increasing amounts of pcDNA-His-AWN004 plasmid DNA (1, 2, and 4 µg) used to transfect cells. Blots were probed with antibodies specific for FLAG, His6, and GAPDH as indicated. (b) The percentages of relative FLAG-UBE2C remaining in each sample were quantitated and are indicated. Data were normalized to the signal for the UBE2C-only control and is expressed as the mean ± SD of biological triplicates. (c) Immunoblots of extracts from HeLa cells transfected with pcDNA-FLAG-UBE2C (2 µg) alone or co-transfected with pcDNA-UBE2C (2 µg) and pcDNA-His-AWN004 or pcDNA-His-AWN004CA (4 µg). (d) Immunoblots of extracts from HeLa cells transfected with pcDNA-FLAG-UBE2C (2.5 µg) alone or co-transfected with pcDNA-UBE2C (2.5 µg) and pcDNA-His-AWN004. Blots were probed with antibodies specific for His6, FLAG, cyclinB1, securin, and GAPDH as indicated. For all immunoblots experiments, the transfected plasmid was supplemented with empty vector. An equivalent amount of total protein was loaded in each lane, as confirmed by immunoblotting with anti-GAPDH antibodies. All immunoblot results are representative of at least three replicate experiments. | |
{kind=link}
To evaluate the degradation effect of AWN004 on endogenous UBE2C, we transfected only the AWN004 into HeLa cells. Unfortunately, the endogenous target ablation was not observed (Fig. S10c in Supporting information). This result may be caused by a variety of reasons including: (ⅰ) the poor transfection efficiency existing delivery system, (ⅱ) the weak binding affinity between our bioPROTAC molecular and target protein [19], (ⅲ) some targets of high clinical relevance can resist degradation by PROTAC [38]. The future work for maximizing the potential of bioPROTACs should focus on developing high affinity binder, such as peptide and nanobody [39,40], or improved gene delivery system [41,42].
In conclusion, we describe the development of a new series of bioPROTACs against UBE2C, a target thought as “undruggable” owing to its lack of deep pockets. The data reported here support that the bacterial-derived NEL domain is a valid candidate to fucntion as the E3 ligase modult in bioPROTAC design. In spite of starting from unimpressive target-binding affnity of initial molecule, natural binder-based bioPROTACs could be optimized by systematically varying the linkers with different length and sequence. Through a series of in vitro and in vivo experiments, we further qualify AWN004 as a potent tool that will be valuable to explore the biology and therapeutic potential of degrading UBE2C.
It should be pointed out that although bioPROTACs can turn into powerful tools and have the potential to become therapeutic agents, their reliance on genetic coding dictates that delivery and drug delivery are key hurdles to overcome. Continued advances in nanoparticle drug delivery methods and viral vector gene delivery systems will play a major part in bringing these novel modalities to patients.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21907006 to Yuxin Ye), the Natural Science Foundation of Guangdong Province (No. 2020A1515011544 to Yuxin Ye) and the Shenzhen Science and Technology Innovation Committee (No. JCYJ20180302150357309 to Yuxin Ye, and No. JCYJ20200109140401752 to Hao Huang).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.08.012.
| [1] |
Y. Lin, W.C. Hwang, R. Basavappa, J. Biol. Chem. 277 (2002) 21913-21921. DOI:10.1074/jbc.M109398200 |
| [2] |
Y. Okamoto, T. Ozaki, K. Miyazaki, et al., Cancer Res. 63 (2003) 4167-4173. |
| [3] |
C. Alfieri, L. Chang, Z. Zhang, et al., Nature 536 (2016) 431-436. DOI:10.1038/nature19083 |
| [4] |
L. Chang, Z. Zhang, J. Yang, S.H. McLaughlin, D. Barford, Nature 522 (2015) 450-454. DOI:10.1038/nature14471 |
| [5] |
L.F. Chang, Z. Zhang, J. Yang, S.H. McLaughlin, D. Barford, Nature 513 (2014) 388-393. DOI:10.1038/nature13543 |
| [6] |
Q. Li, L. Chang, S. Aibara, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 10547-10552. DOI:10.1073/pnas.1607147113 |
| [7] |
Z. Shen, X. Jiang, C. Zeng, et al., BMC Cancer 13 (2013) 192. DOI:10.1186/1471-2407-13-192 |
| [8] |
R. Wang, Y. Song, X. Liu, et al., Int. J. Oncol. 50 (2017) 1116-1126. DOI:10.3892/ijo.2017.3880 |
| [9] |
I. Presta, F. Novellino, A. Donato, et al., Int. J. Mol. Sci. 21 (2020) 1115. DOI:10.3390/ijms21031115 |
| [10] |
M.D. Stewart, T. Ritterhoff, R.E. Klevit, P.S. Brzovic, Cell Res. 26 (2016) 423-440. DOI:10.1038/cr.2016.35 |
| [11] |
A. Poso, J. Med. Chem. 64 (2021) 10680-10681. DOI:10.1021/acs.jmedchem.1c01126 |
| [12] |
M. Bekes, D.R. Langley, C.M. Crews, Nat. Rev. Drug Discov. 21 (2022) 181-200. DOI:10.1038/s41573-021-00371-6 |
| [13] |
M. Pettersson, C.M. Crews, Drug Discov. Today Technol. 31 (2019) 15-27. DOI:10.1016/j.ddtec.2019.01.002 |
| [14] |
M.B. Ludwicki, J. Li, E.A. Stephens, et al., ACS Cent. Sci. 5 (2019) 852-866. DOI:10.1021/acscentsci.9b00127 |
| [15] |
S. Lim, R. Khoo, K.M. Peh, et al., Proc. Natl. Acad. Sci. U. S. A. 117 (2020) 5791-5800. DOI:10.1073/pnas.1920251117 |
| [16] |
F. Cong, J. Zhang, W. Pao, P. Zhou, H. Varmus, BMC Mol. Biol. 4 (2003) 10. DOI:10.1186/1471-2199-4-10 |
| [17] |
Y. Su, S. Ishikawa, M. Kojima, B. Liu, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 12729-12734. DOI:10.1073/pnas.2133261100 |
| [18] |
Y. Ma, Y. Gu, Q. Zhang, et al., Mol. Cancer Ther. 12 (2013) 286-294. DOI:10.1158/1535-7163.MCT-12-0650 |
| [19] |
S. Lim, R. Khoo, Y.C. Juang, et al., ACS Cent. Sci. 7 (2021) 274-291. DOI:10.1021/acscentsci.0c01337 |
| [20] |
K.W. Teng, S.T. Tsai, T. Hattori, et al., Nat. Commun. 12 (2021) 2656. DOI:10.1038/s41467-021-22969-5 |
| [21] |
S. Hatakeyama, M. Watanabe, Y. Fujii, K.I. Nakayama, Cancer Res. 65 (2005) 7874-7879. DOI:10.1158/0008-5472.CAN-05-1581 |
| [22] |
P. Zhou, R. Bogacki, L. McReynolds, P.M. Howley, Mol. Cell 6 (2000) 751-756. DOI:10.1016/S1097-2765(00)00074-5 |
| [23] |
X. Li, L. Shen, J. Zhang, et al., Cancer Res. 67 (2007) 8716-8724. DOI:10.1158/0008-5472.CAN-06-3731 |
| [24] |
E. Caussinus, O. Kanca, M. Affolter, Nat. Struct. Mol. Biol. 19 (2011) 117-121. |
| [25] |
L.J. Fulcher, T. Macartney, P. Bozatzi, et al., Open Bio. 6 (2016) 160255. DOI:10.1098/rsob.160255 |
| [26] |
L.J. Fulcher, L.D. Hutchinson, T.J. Macartney, C. Turnbull, G.P. Sapkota, Open Biol. 7 (2017) 170066. DOI:10.1098/rsob.170066 |
| [27] |
M.R. Baltz, E.A. Stephens, M.P. Delisa, Curr. Protoc. Chem. Biol. 10 (2018) 72-90. DOI:10.1002/cpch.37 |
| [28] |
Y. Ye, Y. Xiong, H. Huang, Commun. Biol. 3 (2020) 752. DOI:10.1038/s42003-020-01492-1 |
| [29] |
N.G. Brown, R. Vanderlinden, E.R. Watson, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 5272-5279. DOI:10.1073/pnas.1504161112 |
| [30] |
E.R. Watson, C.R.R. Grace, W. Zhang, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 17280-17289. DOI:10.1073/pnas.1902889116 |
| [31] |
J. Jumper, R. Evans, A. Pritzel, et al., Nature 596 (2021) 583-589. DOI:10.1038/s41586-021-03819-2 |
| [32] |
D.J. Edwards, , V.P. Ronchi, D.R. Todaro, A.L. Haas, J. Biol. Chem. 289 (2014) 34114-34128. DOI:10.1074/jbc.M114.609164 |
| [33] |
A.D. Portnoff, E.A. Stephens, J.D. Varner, M.P. DeLisa, J. Biol. Chem. 289 (2014) 7844-7855. DOI:10.1074/jbc.M113.544825 |
| [34] |
N.D. Udeshi, T. Svinkina, P. Mertins, et al., Mol. Cell Proteomics 12 (2013) 825-831. DOI:10.1074/mcp.O112.027094 |
| [35] |
V. Akimov, I. Barrio-Hernandez, S.V.F. Hansen, et al., Nat. Struct. Mol. Biol. 25 (2018) 631-640. DOI:10.1038/s41594-018-0084-y |
| [36] |
P. Beltrao, V. Albanese, L.R. Kenner, et al., Cell 150 (2012) 413-425. DOI:10.1016/j.cell.2012.05.036 |
| [37] |
A. Stukalov, V. Girault, V. Grass, et al., Nature 594 (2021) 246-252. DOI:10.1038/s41586-021-03493-4 |
| [38] |
I. Gasic, B.J. Groendyke, R.P. Nowak, et al., Cells 9 (2020) 1083. DOI:10.3390/cells9051083 |
| [39] |
H. Zhang, Y. Han, Y. Yang, et al., J. Am. Chem. Soc. 143 (2021) 16377-16382. DOI:10.1021/jacs.1c08521 |
| [40] |
H. Xu, X. Qin, Y. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2001-2004. DOI:10.1016/j.cclet.2021.09.071 |
| [41] |
Q. Bi, X. Song, A. Hu, et al., Chin. Chem. Lett. 31 (2020) 3041-3046. DOI:10.1016/j.cclet.2020.07.030 |
| [42] |
R. Liu, C. Luo, Z. Pang, et al., Chin. Chem. Lett. 34 (2023) 107518. DOI:10.1016/j.cclet.2022.05.032 |