 2023, Vol. 34
2023, Vol. 34 


Staphylococcus aureus (S. aureus) is a Gram-positive bacterium that commonly colonizes the human skin and nasopharynx. Infections caused by S. aureus present a range of clinical manifestations, such as cellulitis, pneumonia, osteomyelitis, endocarditis and sepsis [1,2]. Importantly, recent studies have shown that invasive S. aureus is a lethal pathogen that can cause a high rate of mortality in hospital and community settings in developed and developing countries [3]. Antibiotic treatment is a routine therapy to combat S. aureus infection in clinics. However, antibiotic resistant S. aureus strains are frequently observed in clinical isolates, which is particularly challenging for clinical management. For example, methicillin-resistant S. aureus (MRSA) has emerged as a global health threat [4,5]. Vaccination is an attractive option to control and prevent bacterial infections [6]. Several anti-S. aureus vaccine platforms, including recombinant proteins derived from S. aureus surface antigens, glycoconjugates composed of capsular polysaccharides, inactivated whole cell and nucleic acid vaccines, have been extensively investigated in the pharmaceutical industry and academia [7]. Some have advanced into late-stage clinical trials but no vaccine has achieved success so far. Thus, developing a vaccination strategy against S. aureus remains a challenge.
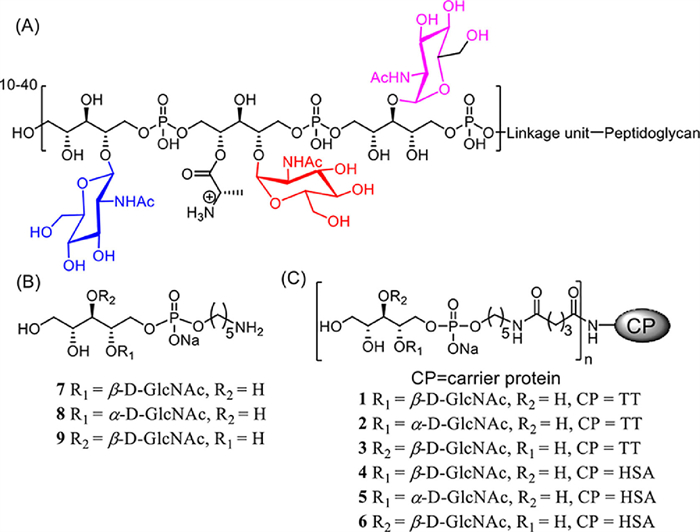
Wall teichoic acids (WTAs) are the major components of the S. aureus cell envelope, which are negatively charged glycopolymers covalently anchored to the peptidoglycan layer of the cell wall [8]. These cell surface polysaccharides are glycosylated ribitol phosphate (RboP) polymers containing 20-40 repeating subunits (Fig. 1A). Chemical structure analyses have revealed that the WTA ribitol residues are modified by d-alanine and three patterns of N-acetylglucosamine (GlcNAc) glycosylation, including α-1,4-GlcNAc, β-1,4-GlcNAc and β-1,3-GlcNAc, where the GlcNAc modifications are completed by three specific glycosytransferases, such as TarM, TarS and TarP, respectively [9-11]. The GlcNAc modification of WTAs on S. aureus play multiple roles in bacterial growth, adhesion and colonization to host cells, biofilm formation, and resistance to antibiotic treatment [12]. Recent studies have shown that host receptors expressed on specific immune cells recognize GlcNAc-modified WTAs and induce the maturation of immune cells to produce proinflammatory cytokines that help the human immune system clear the pathogen. For example, mannose-binding lectin present in human serum recognizes β-1,4-GlcNAc-modified WTAs, which triggers the complement system to destroy S. aureus through an opsonic phagocytosis mechanism [13-15]. Immunological analyses from different groups using bacterial or synthetic glycosylated WTA mimics as models have identified that a substantial amount of antibodies against β-1,4-GlcNAc, β-1,3-GlcNAc and α-1,4-GlcNAc-modified WTA are present in pooled human serum, suggesting that glycosylated WTAs on S. aureus are the dominant antigens that elicit the adaptive immune response in an infectious condition [16,17]. In addition, the isolated serum anti-WTA human IgG antibodies retained the biological function of triggering classical complement-dependent opsonophagocytosis against S. aureus [18,19]. Furthermore, direct immunization with purified WTAs from an S. aureus strain in mice elicits anti-WTA immune response that displays protective immunity against MRSA infection [11,20]. Taken together, these studies suggested that GlcNAc-modified WTAs are promising antigens for developing a vaccine to control S. aureus infections.

|
Download:
|
| Fig. 1. (A) Structure of S. aureus WTA; (B) designed core structures of GlcNAc modified WTAs 7-9 as antigens and their protein conjugates 1-6 (C). | |
{kind=link}
In the present study, three core structures of glycosylated WTAs, including β-1,4-GlcNAc, α-1,4-GlcNAc, and β-1,3-GlcNAc-modified ribitol phosphates 7-9 (Fig. 1B), were designed and chemically synthesized. They were further attached with a pentanyl amine group as a linker, allowing for conjugation with carrier protein tetanus toxoid (TT) to form glycoconjugates 1-3 (Fig. 1C) as anti-S. aureus vaccine candidates. TT was chosen as the carrier protein because it is a highly immunogenic stimulant that is widely applied as a hapten-carrier for developing glycoconjugate vaccines [21,22]. In addition, the corresponding WTA-HSA conjugates 4-6 were synthesized and used as capture reagents for immunologic analyses. The immunological activities of glycoconjugates 1-3 were evaluated in a mice model and the opsonophagocytic killing (OPK) activities induced by antisera of the conjugates were investigated in vitro.
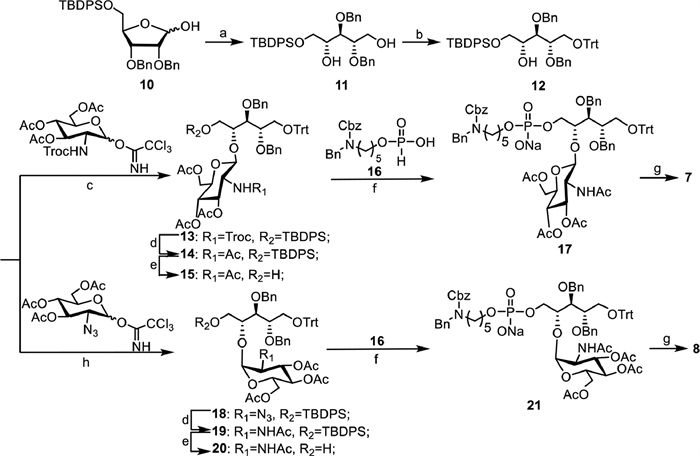
The synthesis of β-1,4-GlcNAc and α-1,4-GlcNAc-modified ribitol phosphates 7 and 8 containing the linker is outlined in Scheme 1. The intermediate 10 was synthesized in four steps starting from commercially available d-ribose according to a previous report [23], which was further reduced using sodium borohydride to provide primary alcohol 11. After selectively protecting the primary alcohol with trityl (Trt) group, acceptor 12 was obtained with a good yield. For the synthesis of β-1,4-GlcNAc-modified ribitol antigen, compound 12 was glycosylated with 2,2,2-trichloroethoxycarbonyl (Troc) protected peracetylated glucose amine Schmidt donor in catalytic amount of TMSOTf to give 13 in 95% of yield. The use of Troc as a neighbor participating group ensured the stereoselective formation of the desired β linkage, which was confirmed by the anomeric proton chemical shift and coupling constant (δ 4.77, d, J = 8.9 Hz). After subsequent transformation of the Troc group to NHAc and removal of the TBDPS group, 15 was obtained in good yield. The condensation of 15 with H-phosphate linker 16 was catalyzed by pivaloyl chloride, followed by in situ oxidation using iodine in one-pot, which proceeded smoothly to provide 17 with a yield of 69% over two steps. Then global deprotection, including removal of the acetyl group with MeONa in MeOH and subsequent hydrogenation to remove the benzyl and Cbz protecting groups with 10% Pd(OH)2/C catalyst, gave final product 7. To synthesize the α-1,4-GlcNAc-modified ribitol antigen, the glycosylation of 12 with a neighbor non-participating 2-azido-2-deoxy peracetylated glucose amine Schmidt donor was performed in a solvent of DCM and diethyl ether (2:1) catalyzed with TMSOTf, where α-selective glycosylation product 18 (α:β = 6:1) was obtained with 85% yield. Then, compound 8 was obtained in a good yield in five steps following similar protecting group manipulations.

|
Download:
|
| Scheme 1. Synthesis of β-1,4-GlcNAc and α-1,4-GlcNAc-modified ribitol phosphates 7 and 8. (a) NaBH4, MeOH, 50 ℃, 30 min, 95%; (b) TrtCl, DMAP, pyridine, 80 ℃, overnight, 79%; (c) TMSOTf, 4Å MS, DCM, −20 ℃, 2 h, 95%; (d) Zn, AcOH, Ac2O, THF, 3 h, 85%; (e) TBAF, AcOH, THF, 40 ℃, overnight, 98% for 15, 93% for 19; (f) PivCl, pyridine, I2, r.t., 3 h, 69% for 17 and 21; (g) MeONa, MeOH, H2, Pd(OH)2/C, H2O, 75% for 7, 65% for 8; (h) TMSOTf, 4Å MS, DCM/Et2O (2:1), −78 ℃, 2 h, 85%. | |
{kind=link}
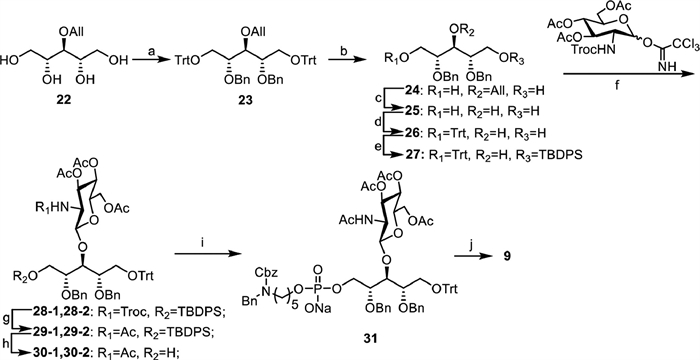
The synthesis of β-1,3-GlcNAc-modified ribitol phosphate 9 containing the linker is shown in Scheme 2. The intermediate 3-O-allyl-d/l-ribitol 22 was prepared in three steps following a reported procedure [24], in which the primary alcohols were selectively protected by Trt and the remaining hydroxyl group was protected with benzyl ether, leading to fully protected ribitol 23. Then, the Trt and allyl protecting groups were removed to deliver alcohol 25 in two steps. After sequential protection of the primary alcohols in 25 with TrtCl and TBDPSCl, the C3-OH ribitol building block 27 was obtained with a good yield. A Troc protected peracetylated glucose amine Schmidt donor was used to couple with 27 under glycosylation condition. Interestingly, a mixture of two inseparable glycosylation products, 28-1 and 28-2, was obtained at a ratio of 1:1 with a 92% yield. However, after transforming the Troc group to acetyl amine and removing of the TBDPS group, compounds 30-1 and 30-2 were successfully separated, and were fully characterized by 1H, 13C NMR and high-resolution mass spectroscopy (HRMS). Both of HRMS spectra displayed identical m/z mass peaks consistent with the calculated glycosylation product. The NMR analysis shows that the anomeric proton signals of 30-1 and 30-2 presented at 5.18 and 5.80 ppm with coupling constants of 8.8 and 8.7 Hz, respectively (NMR data in Table S1 in Supporting information). This result suggests that both of them were β-glycosylation products, which was confirmed by the correlated spectroscopy (1H-1H COSY) and heteronuclear single quantum coherence (HMQC) analyses. Compounds 30-1 and 30-2 were treated with acetic acid to remove the Trt protecting group, separately, and the deprotection products displayed the same 1H, 13C NMR and HRMS spectra. Taken together, these results demonstrate that compounds 30-1 and 30-2 were separable rotamers due to the bulky Trt group in the structure that results in a high barrier to hinder rotation of the single bond. This result is consistent with a recent study in which a ribitol derivative was in a dynamic equilibrium of staggered conformations based on an NMR and MD simulation investigation [25]. Compounds 30-1 and 30-2 were combined, subjected to condensation with H-phosphate linker 16, following global deprotections according to an established procedure to afford compound 9 in five steps with good yield.

|
Download:
|
| Scheme 2. Synthesis of β-1,3-GlcNAc modified ribitol phosphates 9. (a) TrtCl, DMAP, pyridine, BnBr, NaH, DMF, 65%; (b) HCl, MeCN, r.t., 1 h, 91%; (c) PdCl2, MeOH, overnight, 91%; (d) TrtCl, DMAP, pyridine, 80 ℃, overnight, 72%; (e) TBDPSCl, imidazole, DCM, r.t., 1 h, 88%; (f) TMSOTf, 4Å MS, DCM, −20 ℃, 2 h, 95%; (g) Zn, AcOH, Ac2O, THF, 3 h, 97%; (h) TBAF, AcOH, THF, 40 ℃, overnight, 93%; (i) PivCl, pyridine, I2, r.t., 3 h, 78%; (j) MeONa, MeOH, H2, Pd(OH)2/C, H2O, 70%. | |
{kind=link}
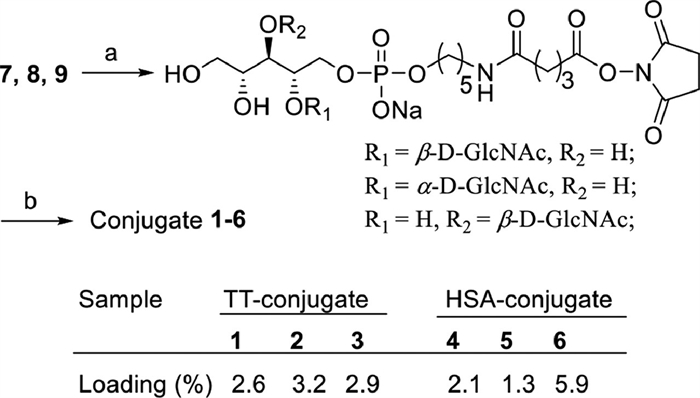
Finally, GlcNAc-modified WTA antigens 7-9 were conjugated to TT or HSA via the bifunctional glutaryl ester method [26]. Briefly, 7-9 were reacted with a large excess (15.0 equiv.) of disuccinimidyl glutarate in a solution of DMF and phosphate-buffered saline (PBS) (4:1) to afford the corresponding activated monoesters (Fig. 2), respectively, followed by a reaction with TT or HSA protein in PBS to generate conjugates 1-6 after purification with a centrifugal filtering device (10 kDa). The antigen loading percentages of glycoconjugates 1-6 were determined by the matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) (Fig. 2 and Fig. S1 in Supporting information).

|
Download:
|
| Fig. 2. Synthesis of conjugates 1-6. Reagents and conditions: (a) disuccinimidyl glutarate, DMF:PBS (4:1), 5 h, r.t.; (b) TT/HSA, PBS, r.t., 3 days. | |
{kind=link}
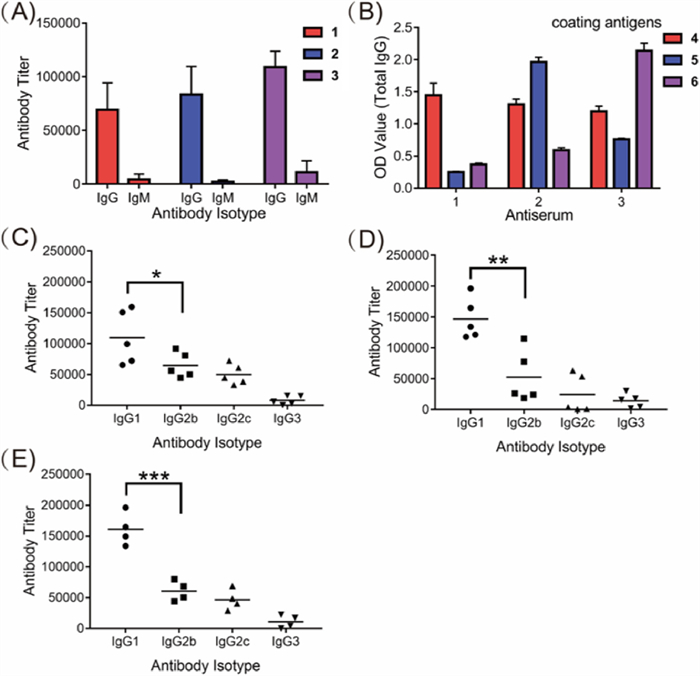
The immunological properties of glycoconjugates 1-3 were evaluated in female C57BL/6 mice. The glycoconjugates (3 µg of glycosylated WTA/mouse/dose) were mixed with Freund's complete adjuvant (CFA) and injected intramuscularly (i.m.) into the mice, followed by three boosts on days 7, 14 and 21 by subcutaneous (s.c.) injection. The baseline blood samples of each mouse were collected from the leg vein on day 0 before the initial immunization. Antisera were prepared from clotted blood samples according to standard protocols and analysed by enzyme-linked immunosorbent assay (ELISA) with corresponding HSA conjugates 4-6 as capture reagents to detect the WTA-specific antibodies. As results, the anti-GlcNAc-modified WTA antibody titers increased gradually as the immunization progressed (Table S3 in Supporting information), and reached the highest level of antibody production after the fourth boost. As depicted in Fig. 3A, the total IgG antibody titers in the mouse antisera elicited by vaccine candidates 1-3 on day 28 were 69165, 83478 and 108886, while a very small amount of IgM antibodies were produced. The IgG isotope was also determined by using corresponding HSA conjugates 4-6 as capture antigens and goat anti-mouse IgG1, IgG2b, IgG2c, and IgG3 as secondary antibodies.

|
Download:
|
| Fig. 3. (A) ELISA results of total IgG and IgM antibody titers in the groups (conjugates 1 and 2 vaccination group, n = 5; conjugate 3 vaccination group, n = 4); (B) Cross-reactivity of total IgG antibodies in the pooled day 28 antisera of conjugates 1-3 with glycosylated WTA haptens in the form of HSA conjugates 4-6 used as coating antigens. ELISA results of the day 28 antisera from individual mice immunized with conjugates 1 (C), 2 (D) or 3 (E). The titers of the corresponding oligosaccharide-specific IgG1, IgG2b, IgG2c, and IgG3 antibodies are displayed. Error bars represent SD of three parallel experiments. *P < 0.05; **P < 0.01; ***P < 0.001. | |
{kind=link}
The analyses revealed that all of the conjugates provoked high IgG1, IgG2b, and IgG2c antibodies titers but a relatively low level of IgG3 antibodies (Figs. 3C-E). These results indicate that GlcNAc-modified WTA 7-9 displayed high immunogenicity and all conjugates provoked strong and specific T cell immunity against GlcNAc-modified WTA in vivo. Using HSA conjugates 4-6 as capture antigens, the cross-reactions of the antiserum elicited by glycoconjugated 1-3 were examined by ELISA. As shown in Fig. 3B and Table S4 (Supporting information), the antisera raised from conjugate 1 did not cross-react with the other two antigens, whereas antisera from conjugates 2 and 3 displayed low cross-reactivity with coating conjugate 4, indicating that the antibodies elicited by conjugates 2 and 3 partially recognized the β-1,4-GlcNAc-modified ribitol antigen 7 structure. This result is slightly different from previous observation where antisera raised from β-1,3-GlcNAc-modified WTAs had strong cross-reaction with β-1,4-GlcNAc-modified WTAs. It is speculated that the slightly different cross-reactivity profile was caused by the different WTA structures as antigens for immunization [18,27]. Our results also suggested that it is possible to produce specific antibodies that can recognize the S. aureus WTAs with different GlcNAc modifications.
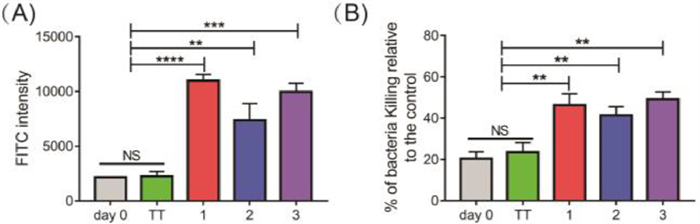
To explore whether the antisera derived from mice immunized with conjugates 1-3 recognized the S. aureus strain, the binding profile of the antisera was examined by fluorescence-activated cell sorting (FACS) technology. We selected Staphylococcus aureus subsp. aureus ATCC 29213 as the model bacterium, which is widely used as a standard quality-control in laboratory testing and is sensitive to antimicrobials, including methicillin [28]. The S. aureus cells were incubated with day 0 (negative control) and day 28 sera, respectively. Then, fluorescein isothiocyanate (FITC)-labelled goat anti-mouse IgG antibody was used to stain the antiserum-treated cells, followed by the FACS analysis. As shown in Fig. 4A, S. aureus cells treated with day 28 antiserum from conjugates 1-3 exhibited significantly higher FITC signals than cells treated with day 0 or TT sera. The binding activities of the antisera from conjugates 1 and 3 were comparable, but significantly stronger than that of conjugate 2. This result indicates that antibodies elicited by vaccine candidates 1-3 were able of recognizing S. aureus cells.

|
Download:
|
| Fig. 4. (A) FACS assays of the binding between S. aureus ATCC 29213 treated with precursor and mouse sera. (B) Opsonophagocytic activities of antisera from each group were measured using the OPK assay. Error bars represent the SD of three parallel experiments. NS, not significantly different, **P < 0.01, ***P < 0.001, ****P < 0.0001. | |
{kind=link}
The in vitro OPK assay was performed to probe whether the antibodies in sera induced by conjugates 1-3 continued their bacterial killing activity mediated by complement system [29]. In these experiments, prefixed S. aureus ATCC 29213 cells were incubated with different dilutions of day 28 antisera of 1-3, followed by the addition of rabbit complement and human-derived polumorphonuclear leukocytes. Thereafter, the S. aureus cells were subjected to colony-forming unit (CFU) assays, where OPK efficiency was calculated based on the bacterial colonies that formed. As depicted in Fig. 4B, the day 0 or TT antisera did not have any opsonophagocytic activity. In contrast, the antisera of all three conjugates displayed significant opsonophagocytic activity, and more than 40% of the S. aureus cells were cleared. This result is consistent with the antibody binding experiments, and demonstrates that conjugates 1-3 are promising vaccine candidates to prevent and control S. aureus infection.
In summary, the core structure of glycosylated WTAs derived from S. aureus, including α-1,4-GlcNAc, β-1,4-GlcNAc and β-1,3-GlcNAc-modified ribitol phosphates were chemically synthesized in a highly convergent and effective way. They were subsequently coupled with TT to generate anti-S. aureus vaccine candidates. In vivo immunological studies demonstrated that glycoconjugates 1-3 provoked robust T cell-dependent responses and elicited high levels of specific IgG antibodies against the glycosylated WTA epitope. We verified that antisera of 1-3 recognized WTAs on MRSA ATCC29213 cells. Further in vitro studies demonstrated that the antibodies elicited by the glycogonjugates mediated remarkable opsonophagocytic killing activity against S. aureus. Taken together, we demonstrated that glycosylated WTAs derived from the surface of S. aureus are potential targets for the development of an anti-S. aureus vaccine. Further work, including synthesis of more complex glycosylated WTAs and cocktail vaccine candidates formulated from different antigens, are underway in the laboratory and will be reported soon.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21472070 and 22177040), and was partly supported by the 111 Project (No. 111-2-06).
| [1] |
L.S. Miller, J.S. Cho, Nat. Rev. Immunol. 8 (2011) 505-518. DOI:10.1038/nri3010 |
| [2] |
C. Li, H. Shen, S. Wang, et al., Chin. Chem. Lett. 29 (2018) 1824-1828. DOI:10.1016/j.cclet.2018.10.025 |
| [3] |
E.K. Nickerson, T.E. West, N.P. Day, et al., Lancet Infect. Dis. 2 (2009) 130-135. |
| [4] |
F.R. DeLeo, M. Otto, B.N. Kreiswirth, et al., Lancet 375 (2010) 1557-1568. DOI:10.1016/S0140-6736(09)61999-1 |
| [5] |
C.P. Harkins, B. Pichon, M. Doumith, et al., Genome Biol. 1 (2017) 130. |
| [6] |
B.K. Giersing, S.S. Dastgheyb, K. Modjarrad, et al., Vaccine 26 (2016) 2962-2966. |
| [7] |
R.S. Daum, B. Spellberg, Clin. Infect. Dis. 54 (2012) 560-567. DOI:10.1093/cid/cir828 |
| [8] |
J.G. Swoboda, J. Campbell, T.C. Meredith, et al., J. Biol. Chem. 11 (2010) 35-45. |
| [9] |
G.Q. Xia, L. Maier, P. Sanchez-Carballo, et al., J. Biol. Chem. 285 (2010) 13405-13415. DOI:10.1074/jbc.M109.096172 |
| [10] |
S. Brown, G.Q. Xia, L.G. Luhachack, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 18909-18914. DOI:10.1073/pnas.1209126109 |
| [11] |
D. Gerlach, Y.L. Guo, C.D. Castro, et al., Nature 563 (2018) 705-709. DOI:10.1038/s41586-018-0730-x |
| [12] |
X. Wu, J. Han, G. Gong, et al., FEMS Microbiol. Rev. 45 (2021) fuaa064. DOI:10.1093/femsre/fuaa064 |
| [13] |
K.H. Park, K. Kurokawa, L. Zheng, et al., J. Biol. Chem. 285 (2010) 27167-27175. DOI:10.1074/jbc.M110.141309 |
| [14] |
D.J. Jung, J.H. An, K. Kurokawa, et al., J. Immunol. 189 (2012) 4951-4959. DOI:10.4049/jimmunol.1201294 |
| [15] |
K. Kurokawa, D.J. Jung, J.H. An, et al., J. Biol. Chem. 288 (2013) 30956-30968. DOI:10.1074/jbc.M113.509893 |
| [16] |
J.H. Lee, N.H. Kim, V. Winstel, et al., Infect. Immun. 83 (2015) 4247-4255. DOI:10.1128/IAI.00767-15 |
| [17] |
J.C. Jung, J.H. Lee, S.A. Kim, et al., Org. Lett. 20 (2018) 4449-4452. DOI:10.1021/acs.orglett.8b01725 |
| [18] |
R.V. Dalen, M.M. Molendijk, S. Ali, et al., Nature 572 (2019) E1-E2. DOI:10.1038/s41586-019-1416-8 |
| [19] |
R. Fong, K. Kajihara, M. Chen, et al., mAbs 10 (2018) 979-991. |
| [20] |
T. Kazue, K. Kenji, M. Patience, et al., PLoS One 8 (2013) 1010-1016. |
| [21] |
S.J. Feng, C.H. Xiong, S.B. Wang, et al., ACS Infect. Dis. 5 (2019) 1423-1432. DOI:10.1021/acsinfecdis.9b00103 |
| [22] |
J. Zhao, G. Hu, Y. Huang, et al., Chin. Chem. Lett. 32 (2021) 1331-1340. DOI:10.1016/j.cclet.2020.10.013 |
| [23] |
H.A.V Kistemaker, G.J. van der Heden van Noort, H.S. Overkleeft, et al., Org. Lett. 9 (2013) 2306-2309. DOI:10.1021/ol400929c |
| [24] |
A.R. Parameswar, P. Pornsuriyasak, N.A. Lubanowski, et al., Tetrahedron 40 (2007) 10083-10091. |
| [25] |
Y. Yamaguchi, S. Ohno, N. Manabe, et al., Molecules 26 (2021) 5471. DOI:10.3390/molecules26185471 |
| [26] |
H. Lin, H.F. Hong, J. Shi, et al., Chin. Chem. Lett. 32 (2021) 4041-4044. DOI:10.1016/j.cclet.2021.04.034 |
| [27] |
S. Ali, A. Hendriks, R. van Dalen, et al., Chem. Eur. J. 27 (2021) 10461-10469. DOI:10.1002/chem.202101242 |
| [28] |
I. Soni, H. Chakrapani, S. Chopra, Genome Announc. 3 (2015) e01015-e01095. |
| [29] |
M. Dwyer, M. Gadjeva, Opsonophagocytic assay, in: M. Gadjeva (Ed.), The Complement System. Methods in Molecular Biology, Vol 1100, Humana Press, Totowa, 2014, 373-379.
|